
Remember me

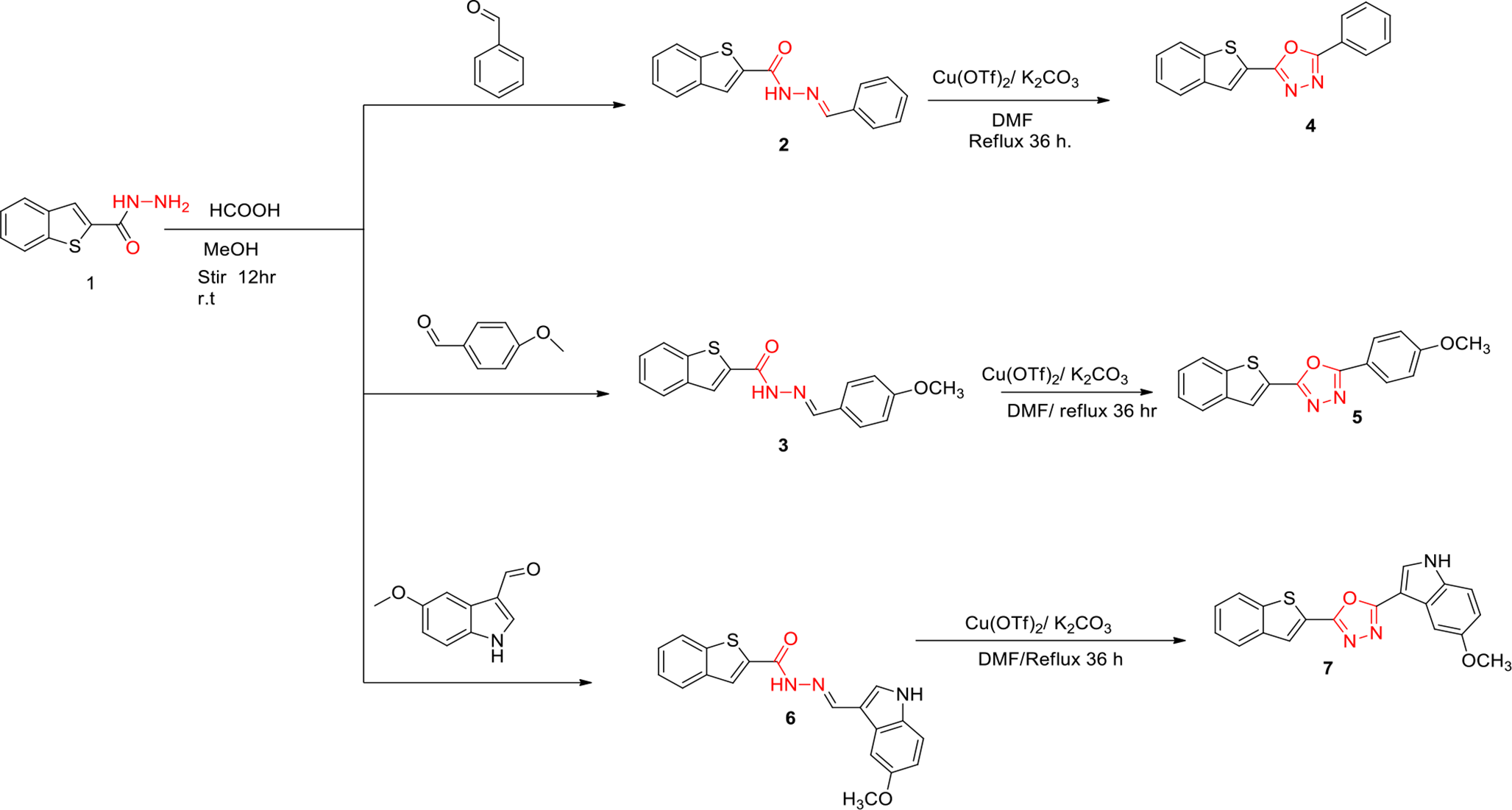
Results will be presented for the temporal-split and macrocycle-split sets for Surflex-Dock, AutoDock Vina, and Gnina. The effects of macrocyclic conformational search adequacy will be explored, as will the degree to which exploitation of prior knowledge affects pose prediction performance.
Fig. 4
Cumulative histograms of temporal split future pose prediction accuracy for Surflex-Dock for the seven most representative individual targets, an example of the CDK2 2XNB top-scoring pose family (top right), and pose prediction accuracy for all 846 ligands (lower right), with red circles indicating performance for top 2 pose families at the 2.0 Å RMSD threshold
Temporal split PINC setThe ligands are spread across ten extensively studied pharmaceutically relevant targets: beta secretase 1 (BACE1), carbonic anhydrase II (CA-II), cyclin-dependent kinase 2 (CDK2), HIV protease (HIV-PR), the non-nucleoside site of HIV reverse transcriptase (HIV-RT), HSP90\(\alpha\), map kinase 14 (MAPK14), PPAR\(\gamma\), protein tyrosine phosphatase 1b (PTP1b), and thrombin (see Methods and Data for additional details). The newly curated set consists of 846 protein-ligand complexes, which was reduced from the original 949 due to elimination of duplicate ligands, removal of ligands from alternative binding sites, and automated quality control in the PDB processing procedure.
The challenge in cross-docking stems from three key issues. First, the protein binding site conformation(s) available for docking may change on binding a ligand that is different from the cognate one, especially if the new ligand is substantially different in structure. Here, because we have employed a 25%-known / 75%-future split, the ligands to be docked contain a large fraction that are very different from the prior known ligands. A second and related issue is that conformational elaboration becomes increasingly challenging, especially for template- and library-based methods, as the structural diversity of the ligands to be docked increases. Because template-based conformational search methods are fit to bound ligand structures that are known at a particular time-point, more recently disclosed molecules present difficulties. Third, binding site definition that is narrowly scoped to prior known binders can bias a docking algorithm against generating even close-to-correct poses, but excessively broadly scoped binding-sites make the search space more challenging.
Figure 4 shows the performance of Surflex-Dock on the full set of ten targets (lower right) and on the seven most representative individual targets. Plots include cumulative histograms for the top 1 (violet), 2 (green), 5 (cyan), and 10 (goldenrod) pose families. The procedure made use of the prior known early ligands during the pose generation process as well as for constructing pose families. Figure 1 shows the full top-scoring pose family for 2XNB-Y8L (lower left). Figure 4 shows the top 20 poses from that pose family. Note that there is some left-to-right variation at the hinge-binding area, with greater heterogeneity seen as the piperazine extends into the solvent (left side). Qualitatively, this type of variation is common across the different targets, with strong protein-ligand interactions (e.g. well-buried polar contacts) exhibiting low variability within pose families and higher variability as ligands extend toward solvent.
The cumulative histograms reflect the minimum automorph-corrected RMSD value for the full set of poses in each of the top N families to the PDB reference structure (note that alternative performance metrics will be discussed below). For the single top-scoring pose family, performance over the entire set at the 2.0 Å RMSD threshold was roughly 66%, generally varying between 60–75%. For the top 2 pose families (red circles in Fig. 4), overall performance was nearly 80%, varying between 75–90%. Performance increased for the top 5 families and less so for the top 10. Note that for HIVRT, the Top 5 and Top 10 were identical and thus appear as the same line.
As mentioned above, one of the new approaches in Surflex-Dock is to make use of the ForceGen conformational search procedure prior to docking. To test if the conformational search beginning from randomized input conformers was adequate, the RMSD of the best pose from the ForceGen conformer ensembles relative to the respective crystallographic pose was determined for each of the 846 future ligands. Figure 4 (lower right, dashed red curve) shows the cumulative histogram of these RMSD values. Overall, 80% of ligands have matches to the crystallographic pose at the 1.0 Å threshold and 99% at the 2.0 Å threshold. Clearly, conformational search was not a limitation on docking performance. In the vast majority of cases, numerous conformations were present in the input ensembles to docking that were close to the experimentally observed, offering many “shots-on-goal” for alignment and further conformational refinement.
Measurement of conformational sampling adequacy provides an optimistic upper bound on possible docking performance. The RMSD values are calculated for each conformer in a pool by identifying the optimal minimum-RMSD alignment of that conformer to the crystallographic reference ligand, which removes a major aspect of the docking search problem. Also, the reference conformer itself is just a model [41], and it generally has been optimized with respect to density fit at the expense of ligand energetics, and without formal contributions from considerations of protein-ligand interactions. Given any docking scoring function and a means to restrain ligand conformational energy, pose optimization will move away from the crystallographic model, typically by 0.1–0.5 Å RMSD. Such optimization within aligned variant non-cognate binding pockets will generally produce larger shifts. Many apparently close-to-correct conformers will exhibit hard clashes or missed key interactions in their low-RMSD optimal alignments, and local optimization may not bring such conformers into high-scoring close-to-correct final poses, even beginning from RMSD-optimal alignments.
Fig. 5
Cumulative histograms of macrocyclic pose prediction accuracy for Surflex-Dock for the best populated representative targets (left, middle), example of the BACE-1 3DV1 top-scoring pose family (upper right), and pose prediction accuracy for all 128 macrocyclic ligands (lower right), with red circles indicating performance for top 2 pose families at the 2.0 Å RMSD threshold
Fig. 6
Effects of conformational search variation on challenging macrocycles: (top) deep ForceGen search addresses the poor docking of rapamycin (cyan) resulting in an accurate docked pose (magenta), and (bottom) torsionally restrained ForceGen search to require the cis-amide shifts the poor docking of geldanamycin (cyan) to an accurate docked pose (magenta)
Further, scoring functions are not perfect, and clearly incorrect poses may score nominally higher than correct ones. Solutions to the docking problem, especially non-cognate docking, must ensure adequate search to avoid hard failures where no close-to-correct solution is explored. The problem of soft failures, where scoring functions incorrectly rank alternative ligand poses, is also significant. As we will see later, poor cross-docking performance for some methods cannot be rescued by giving the correct answer—the crystallographic pose—as input. Adequate conformational sampling is a necessary condition for high accuracy docking, but it is by no means sufficient.
Fig. 7
Comparison of Surflex-Dock performance under different variations for use of prior knowledge: (top) cumulative histograms of performance; (bottom) differences in the best pose from the top-scoring family for HIV protease PDB code 3MXE
Non-macrocycle/macrocycle split setFor this set, the known structures were all bound non-macrocycles, and the ligands whose bound pose was to be predicted were all macrocycles. The set is much smaller (128 ligands across 13 targets) than for the temporal split, owing to the relative scarcity of deposited macrocyclic protein-ligand complexes (see the Methods and Data section for a detailed breakdown). The targets with the largest number of macrocyclic test ligands were: heat shock protein 82 (HSP82), BACE1, factor 11a (FA-11a), FK506 binding protein (FKBP12/5), HSP90, HCV NS3-4A serine protease (HCVP), and myeloid leukemia cell differentiation protein (MCL1).
One can characterize the conformational complexity of a macrocycle based on its “total flexibility”, which includes the total number of exocyclic rotatable bonds plus the total number of rotatable bonds within the macrocyclic ring systems. In our previous work [34], we established that total flexibility of roughly 21–23 rotatable bonds was tractable, yielding the same conformational search accuracy as for non-macrocycles. For the set curated here, all targets but FKBP12/5 fell within the tractable flexibility range.
Figure 5 shows pose prediction performance for Surflex-Dock for four representative individual targets, the specific example of 3DV1, and performance over the full set. The specific case of 3DV1 exhibited a common motif, where conformational variability within pose families was observed within alkyl macrocyclic linkers as well as more solvent-exposed portions of ligands. Strong protein-ligand interactions (e.g. the polar interactions seen in the 3DV1 case) exhibited less variability. Overall pose prediction performance was very similar to that observed for the non-macrocyclic temporal set, with the docking success rate for the top 2 pose families being slightly over 80% at the 2.0 Å threshold.
Pure conformational search effectiveness was also similar between the two sets (dotted red curve, Figs. 4, 5), with 80% of macrocyclic ligands having matches to the crystallographic pose at the 1.0 Å threshold and 100% at the 2.0 Å threshold. Note that there was no statistically significant difference in conformational search performance between the temporal and macrocyclic sets. Kolmogorov-Smirnov comparison of the cumulative histograms of conformer search accuracy for the two sets yielded \(p > 0.1\).
Fig. 8
Comparison of performance on ensemble docking for Surflex-Dock (left column), AutoDock Vina (middle), and Gnina (right) on the temporal split set (top row) and the macrocycle set (bottom row)
All procedures for protein preparation, binding site alignment/selection, conformational search, and docking procedures were identical in the temporal and macrocycles sets. No special treatment was required for macrocyclic ligands.
Surflex-Dock: Macrocyclic challengesThe issue of conformational search adequacy was introduced above. Overall, as shown in Fig. 5 (red dashed line, lower right), macrocyclic sampling was excellent. However, there were two targets, FKBP12/5 and HSP82, for which the default ForceGen conformational search procedure was not sufficient to support the success rates illustrated for the four targets (and overall) in Fig. 5. Figure 6 illustrates the complexities involved with those two cases.
FKBP12/5 was the only target for which macrocyclic ligand flexibility exceeded what has been shown to be tractable for the ForceGen method in prior work [34]. Using the standard search approach, the docking of rapamycin into the macrocycle-naïve ensemble of protein pockets produced a solution among the top 2 with an RMSD of 3.1 Å. The deepest binding pipecolic acid moiety was correctly placed (cyan versus green conformers, Fig. 6 (top)), but the triene responsible for binding mTOR was not correct [42]. However, making use of the deep ForceGen search variant [36, 42] brought performance for the FKBP12/5 targets up to 100% success for the top 2 scoring pose families at 2.0 Å RMSD.
The case of geldanamycin binding HSP82 (and HSP90) is characterized by an unexpected and energetically unfavorable cis amide in the macrocyclic ring system [43] (see red arrows in Fig. 6 (bottom)). Standard ForceGen search does not discover the cis form, resulting in a top-scoring solution with an RMSD of 3.1 Å. Docking results in cases such as this can be improved by making use of prior knowledge of the unexpected conformational behavior of the amide, using a torsional restraint to force the cis amide conformation. When this is done, the substantial fraction of geldanamycin-like ligands in the HSP82 set then produce accurate top-scoring solutions, as seen in Fig. 6 (bottom).
Table 1 Cumulative proportion of dockings within 2.0 Å RMSD of the crystallographic pose per target for the 3 docking methodsEffects of prior knowledgeWe introduced the idea of systematic exploitation of prior knowledge of bound ligands in the study that described the PINC Benchmark [29]. There were two methods: 1) substructural matches of a ligand to be docked to prior ligands were used during docking to add to the pool of alignments for further refinement using the docking scoring function; and 2) ligand similarity to prior known ligand poses to refine the ranking of pose families. In that work, knowledge to guide alignments improved performance roughly 5–10 percentage points in docking success rates, and knowledge used to refine pose family rankings improved performance roughly 10–15 percentage points.
As summarized in Fig. 3, here we replace the substructure-based alignments with those generated using the eSim approach [35]. The method is fast enough that it adds negligible computational cost, and it is able to produce quality alignments without perfect substructural matches. The eSim method has also replaced the prior ligand similarity approach in the pose family rank-refinement procedure, which relies on the same statistical physics approach introduced previously (additional details are provided in the Methods and Data section) [29].
Figure 7 shows the effects on performance for top-2 pose family success rates (top) along with an example of the typical types of improvements seen using an HIV protease ligand as an example (bottom). Use of eSim-based alignments to add to those derived from protomol matching increased performance by roughly 7 percentage points for the non-macrocyclic temporal set and roughly 3 points for the macrocycle set. The effect of pose family rank-refinement using eSim was larger, with a nearly 10 point increase for the temporal set and a 5 point increase for the macrocycle set.
Note also that the previously reported performance of the prior Surflex-Dock version using both types of prior knowledge (red dotted lines in Fig. 7) was roughly 5 percentage points worse than for the new approach. The collection of Surflex-Dock improvements resulted in an overall increase in docking accuracy, a reduction in the degree to which prior knowledge was required to achieve high accuracy levels, and similar levels of performance for docking-based prediction of macrocyclic ligand binding modes to those for non-macrocycles.
Comparison to alternative methodsFigure 8 shows a comparison of Surflex-Dock, AutoDock Vina, and Gnina on both the temporal and macrocycle sets. Neither Vina nor Gnina performed nearly as well as Surflex-Dock, particularly for the macrocyclic ligands. Using a paired t-test, for all numbers of top-scoring pose families for both the temporal and macrocycle sets, performance for Surflex-Dock was statistically significantly better than either AutoDock Vina or Gnina (\(p \ll 10^\)).
Statistical significance can be high even in the absence of a practically meaningful effect. Here, especially for macrocycles, the performance differences were stark. The Vina approach managed just 40% and 25% success for the temporal and macrocycle sets, respectively, for the top two poses families at 2.0 Å RMSD. For Gnina, performance was roughly 60% and 35%, respectively. The comparable performance for Surflex-Dock on both sets was roughly 80% success. Note that while Fig. 8 shows overall performance, target-by-target data also showed Surflex-Dock performance to be superior on a target-by-target basis. Table 1 summarizes performance of Surflex-Dock, AutoDock Vina, and Gnina in terms of success rate at 2.0 Å RMSD for the top 1, 2, and 5 pose families (temporal set targets above the mid-line and macrocyclic targets below).
Fig. 9
Comparison of top 2 pose family performance for AutoDock Vina beginning from native cognate poses (solid green) for the temporal set (top) and the macrocyclic set (bottom), with the comparison to Vina performance using an agnostic starting conformation performance (dotted green lines) and Surflex-Dock performance (yellow)
Conformational search sufficiencyAs we saw in Figs. 4, 5, the conformational search adequacy for the Surflex-Dock approach was not the bottleneck in terms of docking performance (bottom right, red dotted lines, in both Figures). The approach relied on the ForceGen methodology to go from a memory-free starting conformation to a pool of diverse low-energy conformational minima. For both sets, over 90% of the time, conformers were identified that were 1.5 Å RMSD or better compared with the crystallographic poses.
With the exception of Gnina’s performance on the temporal set having a success rate of about 60% for the top 2 pose families (upper right, Fig. 8), Vina and Gnina performed poorly, with success rates of 25–40% (top 2 pose families at a 2.0 Å RMSD success threshold). To understand the source of the large performance gap, we re-ran the Vina procedure, but instead of beginning from memory-free conformations, the ligands were docked beginning with their bound conformations. Figure 9 shows the difference in performance between the two conditions for the top 2 pose families. There was a substantial increase in nominal performance when Vina docking began from the right answer.
The performance gain on the temporal non-macrocyclic set was roughly 5–10 percentage points within the 1.0–2.5 Å RMSD success thresholds, with the gap highlighted at the 2.0 Å RMSD threshold with red circles. For the macrocyclic set, the performance gains were over 20 percentage points (again highlighted with red circles), suggesting that conformational search adequacy on macrocycles is a particular limitation for AutoDock Vina. Note also that even in this artificially biased control condition, performance was still far below Surflex-Dock for both sets (performance shown in yellow using agnostic initial ligand conformations).
Fig. 10
Comparison of original PDB coordinates with re-fit conformational ensembles and top-scoring docked pose families for 2XNB (top) and 3DV1 (bottom)
Cognate re-docking of macrocyclesThese results parallel a recent study from the developers of the macrocyclic search aspect of Vina [44]. The subject of the study was cognate re-docking of macrocyclic ligands, where ligands were docked beginning from their crystallographic poses. When macrocyclic ring systems were held in their native bound geometries, performance was roughly 20 percentage points better than when macrocyclic flexibility was allowed. Vina’s macrocyclic search functionality degraded docking performance.
Similar results were reported in an early study of macrocyclic docking by Bajorath’s group [45]. They reported cognate re-docking performance for 48 macrocycles using MOE LowModeMD [30] for conformational search (beginning from randomized starting conformers) and MOE’s internal docking method. Overall, at the 2.5 Å RMSD success threshold, they achieved a 52% success rate when docking pre-searched conformational ensembles into the cognate protein binding pockets, roughly matching the 53% reported by the Vina macrocyclic docking study [44] when allowing for ring flexibility.
Note that in the Bajorath group study [45], in 47/48 cases, conformational ensembles contained conformers within 2.5 Å RMSD to the bound crystallographic form, so the LowModeMD search was reasonably effective. The authors suggested that a search protocol be developed for macrocycles that combined the generation of conformational ensembles and subsequent ensemble docking, noting that the identifying bioactive conformations is less challenging than predicting accurate compound binding modes that depend on both conformation and alignment. They noted that a limitation of the docking techniques they studied was the inability to treat extended ring systems in a flexible manner and further optimize conformations during docking.
One of the key aspects of the Surflex-Dock approach, introduced many years ago [23], has been the refinement of ligands poses using all-atom Cartesian optimization. This affects the ability of large molecules to be adapted from initial conformations into those that can fit into a protein binding pocket. In general, scoring functions for docking have necessarily bumpy and rugged energy surfaces that stem largely from repulsive terms to prevent protein-ligand clashes. Surflex-Dock treats input conformer pools with additional flexibility during the docking process, including optimization using internal-coordinate alignment/torsional parameters as well as full Cartesian optimization of atomic coordinates. By doing so, it is possible to ameliorate otherwise unrecoverable problems with close-to-correct docked poses.
Limitations of PDB ground-truthIn this work, following common practice including our own prior work, we have used the deposited PDB coordinates of the ligand as “ground-truth” and have used the first set of coordinates in cases where alternates were present. Figure 10 illustrates the impact of this choice using the 2XNB and 3DV1 examples used throughout the foregoing.
In the case of 2XNB, the deposited PDB structure contained two poses for the ligand Y8L, exhibiting variation in the orientation of the thiazole (methyl pointed left vs. right, see top-left of Fig. 10) as well as the piperazine that extends into solvent. This was discussed in the original PINC Benchmark paper [29], where we speculated that additional conformational heterogeneity may fit the X-ray density better. Subsequent to the original work, we developed an approach to fitting conformational ensembles into X-ray density as an alternative to modeling atomic uncertainty with atom-specific B-factors [41], demonstrating better real-space fits as well as producing lower estimates for bound-ligand strain [46, 47]. The ligand of 2XNB when re-fit into density is modeled better by the collection of six conformers shown in orange (middle top of Fig. 10) than by the two alternate B-factor-weighted conformers in the PDB deposition (left top). The characteristics of the top-scoring docked pose family qualitatively match the xGen conformational ensemble (note that both orientations of the thiazole are present within the top 2 docked pose families).
In the case of 3DV1, the deposited PDB structure contained a single pose for the ligand AR9, with high B-factors on the macrocyclic alkyl linker and the aliphatic left-hand-side (left bottom, lighter blue, Fig. 10). As we have previously noted, variation in macrocyclic alkyl linkers is a commonly observed feature when fitting conformational ensembles into X-ray density [41, 46, 47], and the case of 3DV1 is no exception (bottom middle). As with 2XNB, the 3DV1 ligand ensemble fits the experimentally determined density better than the original B-factor-weighted atomic coordinates. Also, the top-scoring docked pose family exhibits heterogeneity in the same manner as the fitted X-ray ensemble. It is also worth noting that bound strain estimates of the re-fit ensembles for both ligands are significantly lower, by nearly 4 kcal/mol for 2XNB-Y8L and just under 3 kcal/mol for 3DV1-AR9.
We have argued that conformational ensembles are the correct model for bound ligands in X-ray structures, and we believe that a more informative measure of docking accuracy for pose prediction than the traditional approach would measure the concordance of a predicted pose family to an X-ray fitted conformational ensemble. One way to do this would be to compute the automorph-corrected RMSD for each pose within a docked pose family to each conformer of the X-ray fitted ensemble. For each predicted pose, the minimum would be taken as the representative value, and the average over all poses within a family would be reported as the final RMSD. In practice, this would not produce large changes in most cases. However, there has been some degree of controversy in the docking field over the most appropriate measure, and we plan to quantitatively explore this in a future study.
Comments (0)