
Remember me

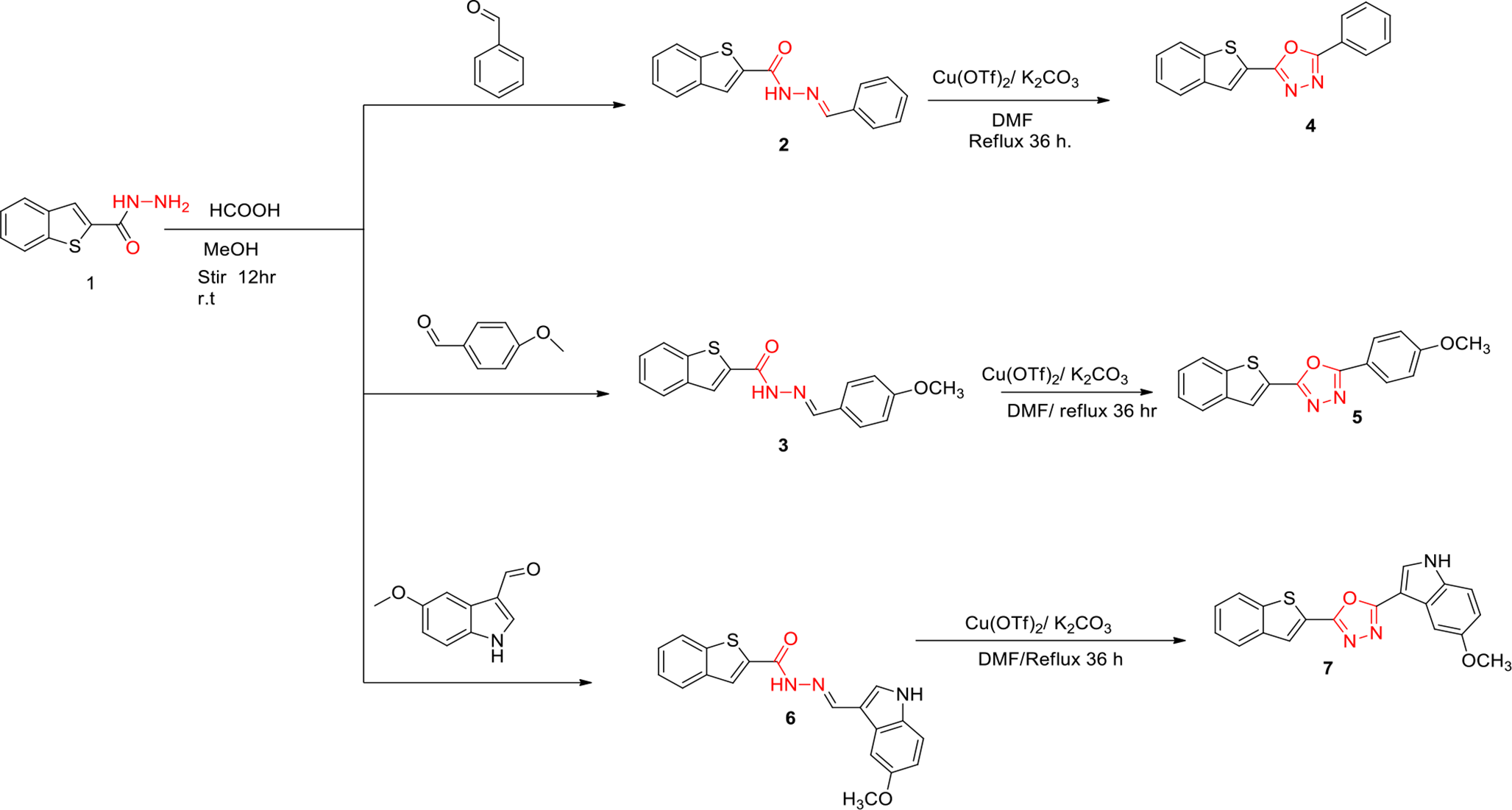
In short, the automated workflow for generating the LOBSTER dataset consists of four steps: First, all ligands are extracted from all PDB files of the RCSB Protein Data Bank (PDB) [37, 38] (Ligand extraction). Second, the ligands are filtered by ligand efficiency (LE) and molecular properties (Ligand selection). Then, ligands binding to identical pockets were superimposed by aligning their binding sites (Ensemble generation). A set of superimposed binding sites with an identical amino acid sequence is denoted as a binding site ensemble [40]. The resulting alignment of ligands is referred to as (ligand) ensemble. Finally, ensembles are grouped into clusters. Only one representative ensemble is picked from each cluster (Ensemble clustering). This fourth step prevents duplicate overlays resulting from redundant subpockets. We will comprehensively describe the four steps of the LOBSTER dataset generation below.
Ligand extractionThe starting point of our workflow is an image of the PDB available on November 7, 2023. Protein structure information, including electron density support, is calculated by StructureProfiler [41]. For all considered ligands, molecular properties, like the number of rotatable bonds or the molecular weight, are calculated using the NAOMI framework [42, 43]. Among others, metal ions and covalent ligands were out of scope for this study and were therefore excluded from the calculations of the NAOMI framework (skipped by construction, \(> 1.5\) Mio. sites). A detailed list of exclusion reasons is provided in the Supplementary Information (SI). A similar processing workflow was utilized for scanning the PDB structures to generate the PDBScan22 dataset [44]. The initial protein-ligand extraction process led to 753,050 binding sites for further consideration.
Ligand selectionIn the next step, drug-like small molecules with a high LE for at least one structure in the PDB are selected. Following the trend [45] to include compounds beyond the Rule of Five [46], we designed less strict filter criteria for drug-likeness to create the largest possible yet adequate dataset. These criteria were derived from multiple sources, as detailed below. An overview of the filtering steps and the number of remaining ligands is provided in Table 2.
Table 2 Filter criteria applied to the ligands and their respective structuresThe only global criterion for the protein structure quality was a resolution of at most 2.5 Å.
The EDIA score [47] quantifies the reliability of the ligand atoms’ fit into the experimental electron density. Only ligands with an \(\textrm_\textrm\) greater than 0.8 were retained, as recommended in the original publication.
Studies on the relationship between size and cell permeability specified an upper limit for molecular weight at \(975\,\hbox ^\) for acceptable permeability [48]. To include this threshold as a filter criterion, molecules with a molecular weight of at most \(\,\hbox ^\) were accepted. A more permissive filter than in other datasets [24, 34] was chosen since the molecular weight of drugs is steadily increasing with the approval of active substances such as natural products and peptides [49, 50].
To exclude lipids and other highly flexible molecules, at most ten rotatable bonds were tolerated inside the molecule following Veber’s Rule for oral bioavailability [51]. Our definition of rotatable bonds matches the definition of Veber et al. (2002): A bond is considered rotatable if it is an acyclic single bond but no amide bond, no bond to a nitrile group, and no bond to a terminal heavy atom.
Molecules with elements other than C, O, N, S, P, Cl, F, Br, I, or B were excluded. Boron, which is not considered a standard organic atom [24], was included due to its increasing appearance in ligands addressing new targets and the tolerability of its toxicity [52].
To exclude most solvents, buffer molecules, and small ions, ligands with less than ten heavy atoms were removed from the dataset. This threshold was previously found to be effective for generating other datasets [24, 53].
In several cases, multiple instances of identical protein-ligand complexes exist. In these cases, only the ligand with the highest \(\textrm_}\), i.e., the best fit to the electron density map, was kept for the respective PDB entry.
The pipeline to find complexes with high LE binders involves an automated parsing of the PDBbind [54, 55] and the BindingDB [56]. Since the PDBbind is a subset of the PDB, there is a direct mapping of the binding affinities or activities to the respective PDB entry and HET code of the ligand. The mapping in the BindingDB is established via sequence comparisons. According to the BindingDB website [57], for a mapping the ligands in both database entries must be identical, and the BindingDB target must have a sequence identity of at least 85 % to the protein chain in the PDB entry. However, to the best of our knowledge no further details for example on the used sequence (ATOM sequence, SEQRES sequence) or the molecular comparison (dissimilarities arising from differences in protonation) are provided. The BindingDB retrieves molecular activity and affinity data from various data sources. We want to emphasize that the choice of data sources might impact the accuracy and comprehensiveness of assay annotations [58, 59]. In particular, the provided metadata is often insufficient to, e.g., reliably exclude pathway-inhibition assays whose readout cannot be attributed to a compound’s activity on a single protein target or cell-based assays that often require tedious post-processing pipelines to exclude false positives (see SI for more details). However, since we are considering both structural and activity/affinity data simultaneously, we are confident that the conclusions on ligand binding are sufficiently robust.
Activity values of the type \(\textrm_\), \(\textrm_\), or \(\textrm_i\) and affinity values of the type \(\textrm_d\) are included. The values are normalized by \(p(}) = -}_(})\). The term to calculate this value is derived from Bento et al. [60]. Please note that we avoid comparing the activity and affinity values from different sources. Instead, we use the values as a common ground to assess the minimum estimated LE of a ligand. Therefore, only the lowest value is used to calculate the LE. For a given value, the formula for calculating the LE [61] of a ligand l with a measured affinity or activity value (\(}_l\)) and heavy atom count (\(}_l\)) is the following:
$$}(l) = \frac}_l)}}_l}$$
Only ligands with a LE exceeding 0.3 are accepted according to the threshold provided by Cavalluzzi et al. [62].
In the last filtering step, the buriedness of each ligand within its protein binding site is calculated with an in-house application to exclude surface binders from the LOBSTER set. Applying the threshold formerly used to generate the sc-PDB [63], ligands that are less than 50% buried by protein atoms are discarded.
In total, the refined selection of compounds comprises 6952 ligands from 6815 protein structures.
Ensemble generation and clusteringTo automatically generate binding site ensembles from our filtered set of ligands, the tool SIENA [40] was utilized. SIENA was applied using the settings specified in the SI. For each ligand, the binding pocket was defined by all residues with at least one atom within 6.5 Å of any ligand atom. Each pocket served as a query for SIENA to search a database of protein structures that contain at least one ligand that passes our filter criteria. All binding sites with identical sequences are extracted. For each query, the resulting binding sites from the SIENA search were superimposed based on the binding site backbone atoms. Thus, the ligands were superimposed indirectly by the alignment of the binding sites. The aligned ligands were extracted as ligand ensembles. For each ensemble, we denote the ligand whose pocket was used for the SIENA search as a search ligand. Initially, 6935 ligand ensembles were generated. If multiple ligands with the same unique SMILES were assigned to the same ensemble, the ligand from the structure with the lowest binding site backbone RMSD was retained. Ensembles containing only one ligand after the deduplication were removed, reducing the number of ensembles to 5836.
Ensuring identical binding site sequences via SIENA, the aligned binding sites most likely also adopt a similar three-dimensional structure. There might be cases with diverging shapes. However, in our opinion, superposition tools should be capable of handling ligand conformations extracted from conformationally flexible binding sites. Thus, conformational flexibility was embraced within the ensemble generation by using the backbone RMSD.
Since differently sized and chemically diverse ligands can bind in partially overlapping sites of a binding pocket, selecting binding residues within 6.5 Å of each ligand will end up in multiple SIENA queries describing different parts of the pocket. To overcome this issue and keep the LOBSTER set as diverse as possible, we used a clustering procedure to retain only non-intersecting binding site ensembles (NBSE) [40]. In this context, non-intersecting means that the same protein-ligand complex should not be part of different ensembles. Otherwise, these ensembles would intersect and thereby describe the same binding pocket. Consequently, ensembles that share at least one protein-ligand complex are grouped into clusters. Algorithm 1 describes the clustering process as pseudocode.
Algorithm 1
Algorithm for grouping intersecting binding site ensembles into clusters. Two ensembles are intersecting if they have a protein-ligand complex in common.
The algorithm depends on the order in which the ensembles are processed. This order affects the number of clusters in the outcome due to the merging procedure described in line 13 and following. By first processing the ensembles with the most ligands, we generate the largest possible number of clusters and, thereby, the largest set of non-intersecting ensembles. If ensembles have the same size, the ensemble name is considered for a tie-break.
Among all ensembles in one cluster, the ensemble resulting from the search ligand with the highest heavy atom count is chosen as a so-called cluster representative. Based on the assumption that larger binding sites may connect several subpockets, this approach ensures larger ensembles. The set of the 671 representatives describes the LOBSTER dataset.
Comparison to approved drugsTo compare the distribution of molecular properties among the compounds in LOBSTER to those of orally available drugs, we used the list of known drugs with an oral route of administration from the FDA Orange Book with at least ten atoms [64]. For this purpose, the compounds from the Orange Book were assigned to structures of approved drugs from DrugBank [65] by name matching (correspondence of the names stored as “GENERIC_NAME”, “JCHEM_TRADITIONAL_IUPAC”, “SYNONYMS”, “INTERNATIONAL BRANDS”, or “PRODUCTS” from DrugBank to either the active ingredient or the proprietary name from the FDA Orange Book). The molecular weight, number of rotatable bonds, SlogP, and the number of hydrogen bond donors and acceptors were calculated with RDKit [66] in KNIME 5.3.0 [67].
Subsets of ligand pairsSince most superposition tools are limited to pairwise alignments [1], we combined ligands belonging to the same ensemble pairwise to extend the scope of the LOBSTER set. One ligand in a pair is considered the template ligand, while the other is called the query ligand. Thus, for two ligands a and b of the same ensemble, both pairs, template a with query b and template b with query a are considered. In an application scenario, the query ligand would be superimposed onto the template ligand. Since the ligand pairs were derived from binding pockets with identical sequences, a high similarity of the pockets is anticipated. Therefore, the ligands’ coordinates are assumed to deliver a reliable and biologically relevant superposition of the ligands. For each pair of molecules, the quality of the overlay is rated by the Shape Tversky Index calculated with RDKit version 2022.09.1 [66]. For a template ligand T and a query ligand Q, the index is calculated as follows:
$$\begin&}(T, Q)\\&\quad = \frac}(T, Q)}}(T, Q) + t \cdot }(T) + q \cdot }(Q)} \end$$
The factors of this equation are chosen \(t = 1\) and \(q = 0\) since we want to evaluate how much of the template molecule is covered by the query molecule. We hypothesize that the closer the index is to one, i.e., the more the query shape overlaps with the template shape, the easier the query can be superimposed onto the template by scoring functions in a way that equals the biologically meaningful alignment. This measure provides an objective assessment of the difficulty of a superposition.
The dataset was split based on the pairwise Shape Tversky Indices with a step size of 0.1, i.e., the set subset_80 contains all pairs with an index of \(0.8 \le x < 0.9\). Of these subsets, subset_90 is considered the easiest since the index of all pairs is at least 0.9.
Calculation of consensus diversityA Consensus Diversity Plot (CPD), as proposed by González-Medina et al. [68], shows the median MACCS fingerprint Tanimoto similarity on the x-axis in ratio to the Scaffold AUC on the y-axis for a given set of molecules. It thereby gives an impression of the diversity in terms of features as calculated through the fingerprint similarity while also showing the novelty of the compounds in terms of the Bemis–Murcko scaffolds. CPDs were generated for the LOBSTER dataset, the subsets generated from LOBSTER, and the AstraZeneca (AZ) dataset [24]. For this purpose, the MACCS fingerprint Tanimoto similarity and the Bemis–Murcko scaffolds were calculated using RDKit [66] version 2022.09.1. For the calculation of the AUC, we utilized scikit-learn [69] version 1.1.3.
Extraction of UniProt and Pfam IDs from LOBSTER and AstraZeneca datasets and protein class assignmentBesides several statistics, a more sophisticated analysis of the protein diversity was conducted. Since we consider the AstraZeneca (AZ) dataset of Giangreco et al. [24] as a previous state-of-the-art dataset for overlays of ligands’ crystal poses, we compared the diversity of proteins from which the ligands were derived for the LOBSTER and the AZ dataset.
In the first step, the exact protein-ligand complexes used by both datasets were extracted. While this step was trivial for the LOBSTER dataset, only the ligands’ three-letter residue names and coordinates are supplied for the AZ set. For PDB entries with multiple ligands with the same residue name, we chose the reference ligand as follows. PyMOL [70] was used to extract all ligands. UNICON [71] was applied to interpret the ligands and add hydrogen atoms. Next, REMUS [72] was used to find the ligand with the lowest RMSD to the one provided in the AZ dataset. In several cases, REMUS could not be applied due to alternate conformations or differences in bond types. Therefore, these entries were manually investigated to find the best matching ligand. The data for this analysis can be found versioned with the LOBSTER dataset in Zenodo. To extract only relevant UniProt accession numbers, the interacting chains as calculated by the StructureProfiler [41] were used to query the PDB Data API [73] for UniProt accession number(s) corresponding to the identifiers of the interacting chain(s). Some PDB entries in the AZ dataset are already obsolete, so these PDB entries had to be substituted by their successors. In addition, the original PDB files were used for both datasets. Original files annotate the chain ID assigned by the author (“auth_asym_id”); the PDB Data API uses the PDB-assigned chain ID (“label_asym_id”). Any altered chains had to be changed accordingly to allow for automated analyses. The resulting data for both datasets can be found in Zenodo as well. For any UniProt accession number, the Pfam ID and the Pfam name were extracted using the Pfam Data API [74]. If no UniProt accession number is provided for an interacting chain, it is omitted from the analysis. In the LOBSTER dataset, the UniProt accession numbers for the entries in eight clusters are not annotated in the PDB. These clusters are omitted from the protein diversity analysis. Notably, all but two UniProt accession numbers annotated in the AZ dataset could be reconstructed using this automated analysis, with the two mismatched UniProt accession numbers no longer being supported (P0A5J2:P9WK19 and P0C5C1:P9WKD3). Protein classes were assigned manually based on Pfam and chain IDs, and a complete table is provided with the LOBSTER dataset. A detailed description of the assignment of protein classes based of Pfam IDs is available, too.
Comments (0)