Experimental animals
Twenty-four male Sprague-Dawley (SD) rats (6-week-old; weighing 257 ± 10 g) were housed in a specific pathogen-free environment. Two rats per cage were maintained in a temperature-controlled room (23 ± 2 °C) with a 12-h light and 12-h dark cycle. Food (standard diet or high-fat diet) and water were provided ad libitum. Daily food intake was recorded, while body weights were collected for every 3-day once. All procedures complied with the National Research Council Guidelines for the Care and Use of laboratory animals. This study was conducted according to the animal management regulations of the Ministry of Health of China, and was approved by the Animal Ethics Committee of Zhejiang Normal University (No. ZSDW2022027).
Animal grouping and treatment
After a one-week adaptation period, the rats were randomly assigned into three groups, including control (CON, n = 8), high-fat diet (HFD, n = 8) and HFD plus aerobic exercise (HE, n = 8), and treated for a period of 6-week. Rats in the control group fed a normal diet obtained from the Research Diets (Research Diets Inc., NJ, USA). The rats in HFD and HE groups fed a 60% high-fat diet (D12492, Research Diets Inc., NJ, USA). Following HFD feeding, the rats in HE group performed an aerobic exercise training for a period of 6-week, as described in the protocol below.
Treadmill exercise protocol
Prior to the actual exercise session, rats were familiarized with the treadmill environment. For familiarization, rats in the HE group ran on a treadmill at low running speed (16 m/min) for 20-minute on day 1. Then the running speed (2 m/min/day) and running time (10-min/day) were gradually increased to reach the target speed (24 m/min) and time (60-min) [22]. The final exercise training protocol consisted of 60-min/day, 5 days/week for a period of 6-week. The exercise load comprised a running of 15 m/min for the first 5-minute, 20–25 m/min for the next 50-minute, and 15 m/min for the last 5-minute, without incline. The exercise program was scheduled from Monday to Friday between 5:00 and 7:00 p.m. The exercise intensity ranged from approximately 60–70% of VO2max [23]. We adopted this exercise protocol based on our previous study, which reported to be effective in producing the physiological changes in SD rats [22].
Oral glucose tolerance test (OGTT) and insulin sensitivity
OGTT was performed twice in our study following a 12-hour fasting period. The first OGTT was performed prior to the intervention (pre or baseline), and the second OGTT was performed after the intervention (post). Following the exercise training, the animals rested for 12-hour (while feeding), and then fasted for 12-hour before the OGTT. Blood samples were collected from the tail of each rat at 0-, 30-, 60-, 90-, and 120-minute after an oral administration of glucose (2 g/kg body weight). The changes in blood glucose levels were measured using a glucometer (Sinocare, Changsha, China), and recorded for three times to calculate the average value. The glucose area under the curve (AUC) was calculated and presented as histograms [24].
Serum insulin levels were measured using the rat insulin ELISA kit (Sangon Biotech, Shanghai, China), according to the manufacturer’s instructions. The Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) was calculated as follows:
HOMA-IR = ((fasting plasma insulin in µIU/mL) × (fasting plasma glucose in mg/dL))/405 [25].
Sample collection and biochemical assays
Forty-eight hours after the last exercise session (6-week), rats were anesthetized with urethane (2 g/kg, intraperitoneally) following a 12-hour fasting period. The fasting time was the same in CON and HFD groups to ensure the similar conditions in all groups. Blood samples (5 mL) were collected, and centrifuged at 3000 rpm for 15-minute. Then, the portions of the quadriceps were excised for different analyses: one portion was embedded in the optimal cutting temperature compound (OCT, Sakura, Tokyo, Japan), and frozen in isopentane, while the other portion was frozen in liquid nitrogen, and stored at -80 °C. The quadriceps muscle was selected for its balanced distribution of type I and type II muscle fibers, ease of access for reliable sampling, and its relevance to metabolic outcomes [26, 27].
In addition, the subcutaneous fat was carefully dissected beneath the dermis, ensuring minimal contamination from surrounding tissues. Following this, epididymal fat was excised from around the epididymis. Special care was taken to preserve its structural integrity. Both fat tissue samples were weighed on an analytical balance (Sartorius, BSA224S, Germany). The weight of each fat depot was recorded immediately to determine the total fat mass for subsequent analysis.
Periodic acid-schiff (PAS) staining and measurement of skeletal muscle glycogen content
For histological analysis, muscle tissue Sect. (10 μm thickness) were prepared using a cryostat (Leica CM1860, Germany). The sections were mounted on glass slides, and fixed with 4% paraformaldehyde for 10-minute at room temperature before undergoing PAS staining. The slides were immersed in periodic acid solution for 10-minute, washed three times with distilled water, and incubated with Schiff solution at 37 °C for 45-minute. Finally, the slides were washed with distilled water for 5-minute, and dehydrated using graded alcohols.
To measure the glycogen levels in the quadriceps, the glycogen assay kit (BC0345, Solarbio, China) was used, and performed according to the manufacturer’s instructions. Briefly, snap-frozen quadriceps (80 mg) were homogenized, and diluted with 1 mL distilled water. The homogenate was boiled at 100℃ for 20-minute, centrifuged at 8000 g for 10-minute at 25℃, and then the supernatant was collected. Glycogen was extracted by a strong alkaline extract solution, and the glycogen content was determined using anthrone chromogen under strong acidic conditions [28].
Quantitative real-time polymerase chain reaction (qPCR)
Quadriceps muscle tissue (50 mg) was lysed with TRIzol reagent to extract total RNA. The concentration and purity of the extracted RNA were assessed with a NanoDrop spectrophotometer (Multiskan SkyHigh, Thermo Fisher Scientific, Waltham, USA). The RNA purity was determined by measuring the absorbance ratios at 260/280 nm and 260/230 nm on a NanoDrop spectrophotometer (Multiskan SkyHigh, Thermo Fisher Scientific, Waltham, USA), ensuring the ratios were above 1.8 and 2.0, respectively, to confirm high purity. For cDNA synthesis, 1 µg of total extracted RNA from each sample was used with the PrimeScript RT Master Mix (Takara, Shiga, Japan). The qPCR assays were performed using PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, Waltham, USA) to ensure specific and sensitive detection of target gene expression. All qPCR analyses were conducted on the CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, USA). The reaction conditions comprised an initial pre-denaturation step at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 10 s and annealing/extension at 60 °C for 30 s. Gene expression levels were quantified using the 2 − ΔΔCt method with GAPDH serving as the internal reference [22]. The primer sequences are provided in Table 1.
Table 1 Primers for qPCR used in this studyWestern blot
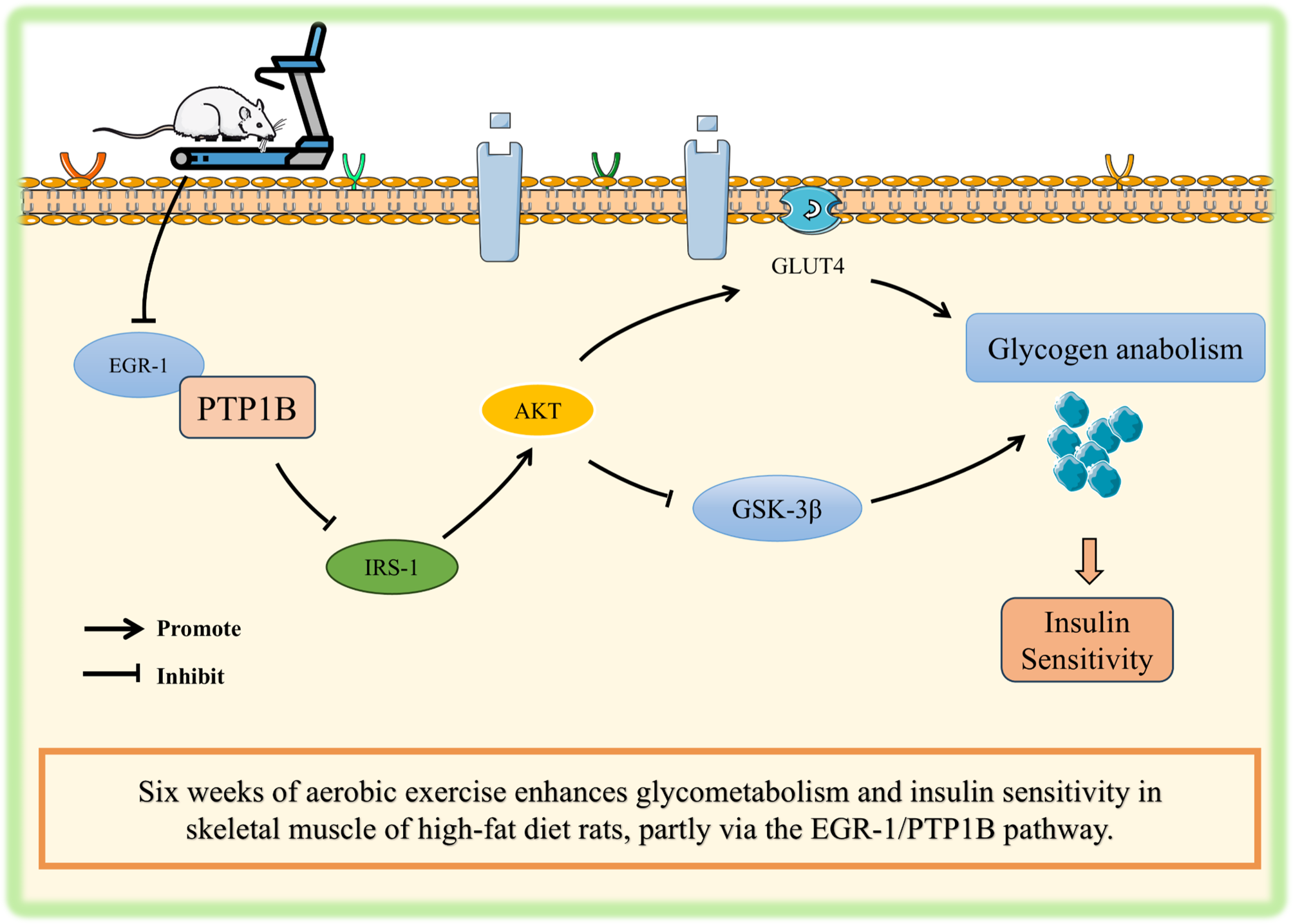
Total protein was extracted from rat quadriceps muscle using the commercially available RIPA lysis buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP40, 1% sodium deoxycholate, and 0.1% SDS, Thermo Fisher Scientific, 89901USA) with protease and phosphatase inhibitors (Thermo Fisher Scientific, 78440, USA). The protein concentration was determined using the BCA protein assay kit (Thermo Fisher Scientific, 23235, USA). Equal amounts of protein along with a protein ladder (Bio-Rad, 1610374, USA), were subjected to SDS-PAGE for electrophoresis. Electrophoresis was conducted at a constant voltage of 70–100 V until the dye front reached an appropriate distance. Following separation, proteins were transferred to polyvinylidene fluoride (PVDF) membranes using a wet transfer system (Bio-Rad) at 80 V for 2 h. The transfer buffer consisted of 25 mM Tris, 192 mM glycine, and 20% methanol. the PVDF membranes were then blocked in Tris-buffered saline with Tween® 20 (TBST, 50 mM Tris, 150 mM NaCl, 0.1% Tween-20, pH 7.4) containing 5% skimmed milk at room temperature for 1 h. The membranes were then incubated overnight at 4 °C with primary antibodies specific for EGR-1 (Proteintech, 22008-1-AP), PTP1B (Santa Cruz, sc-133259), GSK-3β (Proteintech, 67329-1-Ig), phospho-AKT Ser473 (p-AKT, Proteintech, 66444-1-Ig), AKT (CST, 9272), phospho-IRS1 Ser307 (p-IRS1, CST, 2381), IRS1 (Proteintech, 17509-1-AP) and GLUT4 (Proteintech, 66846-1-Ig), all diluted at a ratio of 1:2000. After washing the membranes three times with TBST, they were incubated at room temperature for 2 h with appropriate secondary antibodies: goat anti-mouse (Proteintech, SA00001-1) or goat anti-rabbit (CST, #7074), also diluted at 1:2000. Detection of protein bands was performed using an enhanced chemiluminescence kit (Thermo Fisher Scientific, Waltham, USA) and visualized with a gel imaging system (Bio-Rad, Hercules, USA). The optical density values of the target proteins were quantified relative to that of GAPDH using Fiji software [29].
Immunofluorescence
Muscle samples were collected, and immediately frozen in OCT. Using a cryostat (Leica CM1860, Germany), 10 μm thick sections were cut, and mounted on glass slides. The slides were fixed with 4% paraformaldehyde for 10-minute at room temperature. After three washes with PBS (5-minute each), slides were permeabilized with 0.5% Triton X-100 in PBS for 20-minute, followed by three additional washes with PBS. Subsequently, the slides were blocked with 1% BSA in PBST (PBS + 22.52 mg/mL Glycine + 0.1% Tween-20) for 30-minute at room temperature. Primary antibodies, EGR-1 and PTP1B, were incubated overnight at 4℃. Slides were rinsed three times with PBS, and incubated with fluorescent secondary antibodies (Alexa Fluor 488-conjugated goat anti-mouse, Abcam, AB150113 or Alexa Fluor 594-conjugated goat anti-rabbit, AB150080) at a ratio of 1:500 in antibody dilution buffer for one-hour at room temperature in the dark. Finally, the cell nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (Beyotime Institute of Biotechnology, China). The stained tissue was observed, and photographed using a fluorescence microscope (Leica, Wetzlar, Germany) [29].
Statistical analysis
Data were statistically analyzed using IBM SPSS Statistics 20 (Chicago, USA), and image analysis and mapping were performed using Fiji software and GraphPad Prism 9 (San Diego, USA). The Pearson correlation coefficient of the selected images was analyzed using Fiji software. All data were summarized as mean ± SEM. Comparisons between multiple groups were conducted using a one-way analysis of variance (ANOVA), with post hoc analysis by Tukey’s multiple comparisons test. A significance level of P < 0.05 was considered statistically significant.



Comments (0)