
Remember me




The aim of this randomized, placebo-controlled, double-blind, prospective trial was to examine the efficacy of BlephEx™ treatment for chronic blepharitis by determining whether BlephEx™ treatment improves quality of life and ocular surface parameters in patients with chronic blepharitis compared to sham treatment. Our primary outcome was the change in the Ocular Surface Disease Index (OSDI), a validated metric for quality of life related to dry eye disease and blepharitis. Secondary outcomes included the severity of eyelid inflammation measured by the Efron grading scale for eyelid margin disease, tear break-up time (TBUT), and Schirmer's test to evaluate tear production.
This study aimed to assess whether automated eyelid margin debridement with BlephEx™ provides benefits beyond a sham procedure, which controls for mechanical eyelid stimulation and placebo effects. Patients were randomized to receive either BlephEx™ debridement per the manufacturer’s instructions or sham treatment under equivalent conditions. Outcomes in the BlephEx™ group were compared to those in the sham group to determine the impact of BlephEx™ treatment relative to stimulation and placebo effects alone.
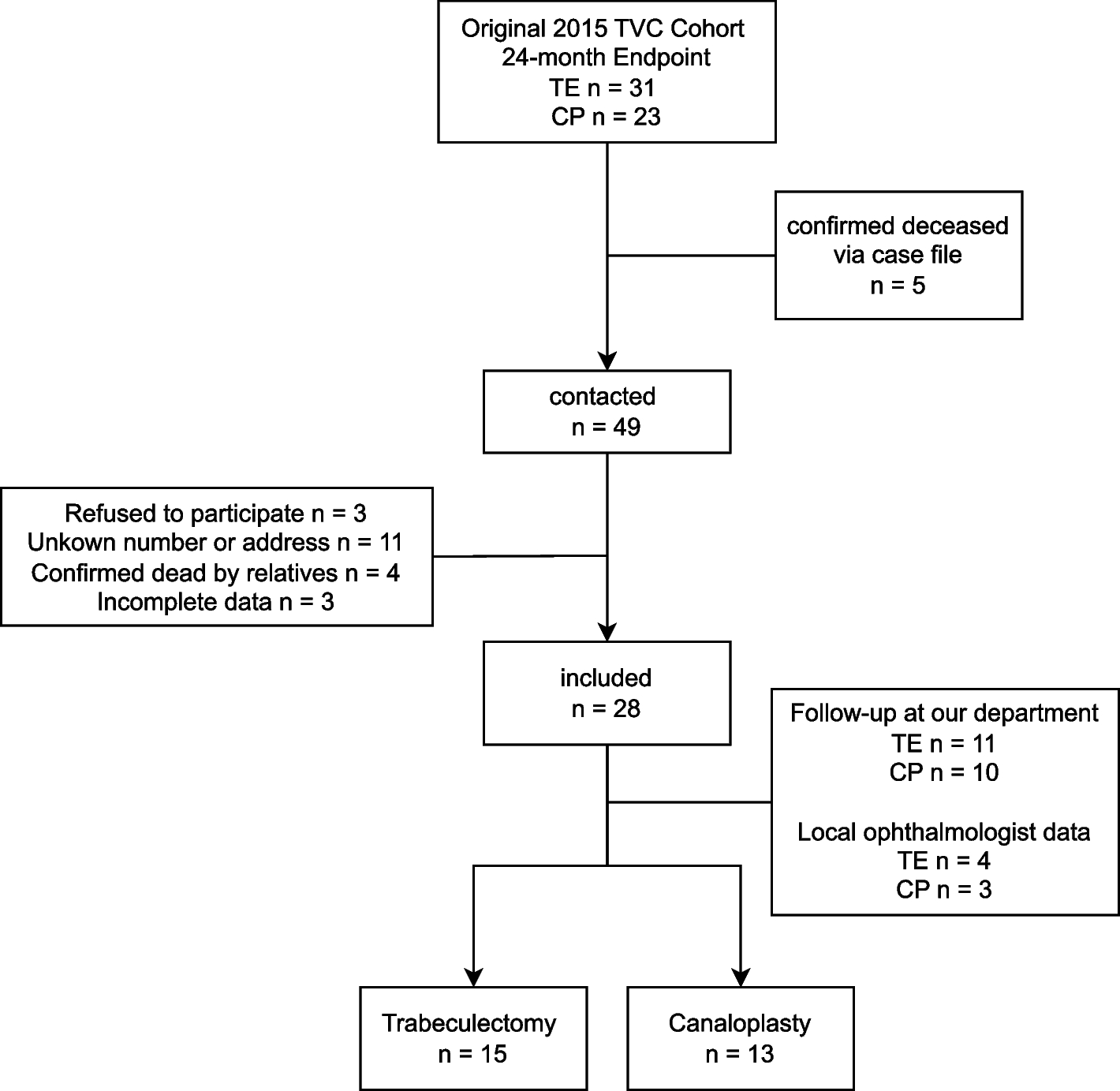
The study was conducted at the Eye Center of the University Hospital Freiburg after approval by the local ethics committee (application no. EK-Freiburg 349/17) and can be accessed in the research database of the University of Freiburg. (https://forschdb.verwaltung.uni-freiburg.de/forschdbukl/rech_frame_projekt.htm as of 02.01.2023). The study conformed to the Declaration of Helsinki. The trial registration was carried out retrospectively on February 16, 2024 in the DRKS (German Clinical Trials Register) at https://drks.de/search/de/trial/DRKS00033492) under the trial registration number DRKS00033492. A crossover design was used as a recruitment aid (study participants received verum treatment in each case) and was not considered for the statistical analysis (see Fig. 2). Therefore, only the baseline (examination before treatment) and the examination 4 weeks after treatment (switch at the time of crossover) visits were evaluated to minimize pretreatment bias. The patients were divided into 2 groups randomized to receive either BlephEx™ or sham treatment. Afterwards, the participants were switched to the alternative intervention. The observed treatment effect in this second stage was not taken into account in the statistical analysis to avoid a potential carryover effect, which could have influenced the results.
Fig. 2
Patient flowchart study: study enrollment and progression
Trial participantsThe inclusion criteria were symptomatic blepharitis (at least Grade 2 according to the Efron grading scale), age older than 18 years, decimal visual acuity > 0.5, blepharitis refractory to eyelid margin care for at least 4 weeks, and the use of lubricating drops.
The exclusion criteria were routine (daily) contact lens wear, other eyelid or ocular surface diseases, previous ocular surgery, previous treatment with cyclosporin A or steroids, pregnancy, autoimmune diseases (e.g., rheumatologic conjunctivitis, Sjögren's syndrome, endocrine orbitopathy) or other immunological diseases and acute infections. Further exclusion criteria were the presence of trachoma, atopic dermatitis, diabetes mellitus, severe systemic diseases, graft-versus-host disease or the use of antibiotics in the last 4 weeks.
The screening process was as follows: At the first contact in our outpatient clinic, patients with blepharitis were screened by the examining doctors according to our inclusion and exclusion criteria. If someone was generally eligible for participation in the study, a member of the study team was contacted. A special medical history form was used to assess all the inclusion and exclusion criteria. If all the criteria for participation in the study were fulfilled, the patients were informed and given a consent form. Subsequently, an appointment was made for the initial examination and treatment. Patients were recruited between December 2017 and June 2018.
In the case of a new occurrence of the disease, basic therapy, which included four weeks of eyelid margin care and lubricating drops, was recommended first. If the symptoms did not subside sufficiently within 4 weeks of treatment, the patient was included in the study.
InterventionsBlephEx™ (Scope Ophthalmics, London, UK) treatment was performed according to the manufacturer’s instructions after topical anesthesia. The cleaning head was placed on the BlephEx™ device (one per eye), and Blephagel® from Théa Pharma GmbH was applied as a cleaning gel (composition: agua, poloxamer 188, PEG-90, carbomer, and sodium hydroxide). Then, the upper and lower lids of each eye were treated for two minutes according to the manufacturer's instructions. The practitioner moved along the eyelid margin and removed both coarse and minor "debris" (see Fig. 1).
The sham treatment was carried out under the same temporal and spatial conditions. The cleaning head was placed on the BlephEx™ device (one per eye), and cleaning gel was applied. Then, each eye was treated for two minutes on the upper and lower lid. However, the doctor did not move along the eyelid margin but rather along the adjacent row of eyelashes on the eyelid skin so that no mechanical cleaning of the eyelid margin could be assumed. Due to the anatomical proximity, the resulting transfer of sensation to the eyelid and the previous superficial anesthesia, it can be assumed that it was not possible for the patient to distinguish between BlephEx™ treatment and sham treatment.
Outcome measuresThe primary endpoint was a change in quality of life measured by the ocular surface disease index (OSDI), as described by Schiffman et al. [13]. The scores at the different time points were compared with each other and statistically analyzed at the end of the study.
The secondary endpoints were the severity of lid margin inflammation, graded according to the Efron grading scale, as well as the evaluation of tear break-up time (TBUT) and the Schirmer test.
The Efron grading scale was used to assess eyelid margin involvement in a blinded manner based on the eyelid margin photos before and 4 weeks after treatment; the assessments were performed by two independent blinded examiners on a scale of 0 to 4. The decisive factors were redness of the eyelid margin, condition of the meibomian gland excretory ducts, incrustations and adhesions on the eyelashes, telangiectasia on the eyelid margin, and visible surrounding skin irritations [14].
The tear break-up time (TBUT) was measured using a 2% fluorescein solution. Fifty microliters of this solution was dropped into the lower fornix with a pipette and distributed over the surface of the eye by blinking. This was followed by examination via a slit lamp in the photo laboratory with blue light. After a short exposure time, the patient was asked to close his eyes, open them on command and then keep them open. This was ensured by the examiner with the help of a cotton swab. The time until the tear film broke up was determined and recorded. The procedure was repeated three times for each eye.
The Schirmer's test II was performed to obtain information on baseline tear production. For this purpose, oxybuprocaine 0.4% eye drops were given, and after an exposure time of five minutes, the Schirmer test strips were hooked on the lateral lower lid. The patients were asked to close their eyes loosely. After 10 min, the test strips were removed, and a ruler was used to directly measure how much fluid the strips had absorbed.
Best-corrected visual acuity was assessed using Early Treatment of Diabetic Retinopathy Study (ETDRS) charts after standardized subjective refraction by trained personnel [15]. VA was converted to logMAR for statistical analysis.
Sample sizeGiven that this was an exploratory approach, with no firm prior knowledge of effect sizes or expected variances, it was not possible to calculate the sample size beforehand.
RandomizationSequence generationThe randomization sequence was generated in advance with the package "blockrand" from the R-platform at a ratio of 1:1 for the verum patients to the control patients and was stored covertly for the study personnel in the database.
Allocation concealment mechanism and implementationRandomization was automated upon study inclusion. The group allocation in combination with a randomization number was directly transmitted electronically to the treating physician only by e-mail. Moreover, it was ensured that this information was not visible to anyone involved in the diagnostic process during the course of the study.
BlindingThe study treatments were performed by an unmasked study team of two ophthalmologists. Diagnostics were performed by a doctoral fellow, who was blinded to the treatment performed until the end of the study. The study team (two ophthalmologists) involved in the assessment of the Efron grading scale was also blinded at all times. All study patients were also blinded regarding the type of treatment (see description of sham treatment above).
Statistical methodsCategorical data are presented as percentages. For the analysis of categorical variables, the Chi-Square Test was applied when the expected frequencies in all categories were above 5. In cases where the expected frequencies in any category were below 5, the Fisher's Exact Test was used to ensure the accuracy of the results. This approach ensures appropriate testing for both large and small sample sizes, depending on the distribution of the categorical data across groups.
All continuous data were tested for normality using the Shapiro–Wilk test. For normally distributed data (e.g., OSDI), the paired t-test was used to calculate the p-value of the change before and after treatment within each treatment group, and the unpaired t-test was used to calculate the p-value for differences between the two treatment groups.
For data that were not normally distributed, the Wilcoxon signed-rank test was applied to calculate the p-value for the change before and after treatment within a treatment group. The Mann–Whitney U test (also known as the Wilcoxon rank-sum test) was used to calculate the p-value for comparing differences between the treatment groups.
For each primary and secondary outcome, results for each group were reported, and the estimated effect size and its precision (95% confidence interval) were calculated in accordance with the Consolidated Standards of Reporting Trials (CONSORT) 2010 checklist. The difference before and after treatment was determined by calculating the mean and 95% confidence intervals, as well as the median and interquartile range (IQR). For normally distributed data, Cohen’s d was calculated to compare the effectiveness of the different treatments. Cohen proposed treating d = 0.2 as a "small" effect size, 0.5 as a "medium" effect size and 0.8 as a "large" effect size. This implies that if the difference between the means of two groups is less than 0.2 standard deviations, the difference is considered to be negligible, even if it is statistically significant.
For non-normally distributed data, the Rank-Biserial Correlation (rbc) was used as the effect size for comparisons made with the Mann–Whitney U test. While there are no universally agreed-upon thresholds for small, medium, or large effect sizes in the context of rbc, a similar framework to that of Cohen’s d was applied, with rbc ≈ 0.10 representing a small effect size, rbc ≈ 0.30 representing a medium effect size, and rbc ≈ 0.50 or higher representing a large effect size.
All statistical analyses were performed using R version 4.3.1 and 4.4.1 (R Foundation for Statistical Computing, Vienna, Austria).
Comments (0)