Reagents and antibodies
Table S1-S4 provides a comprehensive description of the antibodies, primers, plasmids, siRNAs, chemicals and cell lines used in this study.
Cell culture
BMSCs were obtained from the femurs and tibias of 6-week-old SD rats following established procedures [16]. The cells were maintained in α-MEM medium (Cyagen, China) supplemented with penicillin, streptomycin, and fetal bovine serum (FBS) (Cyagen, China) at 37 °C. Cells in passages 3 to 6 were used for all experiments. The medium was refreshed every 3 days, and cells were passaged when they reached 80% confluence. A DMEM-based medium was used to culture RAW264.7 cells supplemented with 10% FBS, 1% penicillin/streptomycin, and 50 ng/mL RANKL.
Osteogenic induction
Forty-eight hours after seeding, BMSC osteogenic differentiation was induced. The osteogenic differentiation medium was purchased from Cyagen Biosciences (Soochow, China). The medium was exchanged every 3 days.
Adipogenic induction
Adipogenic differentiation medium was purchased from Cyagen Biosciences (Soochow, China). When the BMSCs had propagated to a density of 100%, the culture medium was switched to adipogenic differentiation medium A, and after 3 days of intramuscular culture, the medium was replaced with adipogenic differentiation medium B. The BMSCs were then maintained in medium B for 24 h. The two media were altered 3–5 times, and then the BMSCs were maintained with medium B, which was changed every 2–3 days until large lipid droplets were apparent.
CCK8 assay
We plated BMSCs (1 × 103 cells/well) in 96-well plates and then cultured them in a medium containing a variety of chemical concentrations. Then, A fresh medium containing 10% CCK8 stock solution (Beyotime Biotech, China) was added to the cells and incubated at 37 °C. We measured the absorbance at 450 nm after two hours.
Western blotting
The protein isolation protocols were performed following our previous publication [4]. Briefly, the cell lysates were subjected to electrophoresis and transferred onto nitrocellulose membranes. The membranes were blocked using QuickBlock Buffer (Beyotime Biotech) for 1 h. Subsequently, the membranes were incubated overnight at 4 °C with primary antibodies. After washing thrice with PBS, the membranes were exposed to the relevant secondary antibodies and detected through chemiluminescence using Pierce ECL. The resulting autoradiograph was analyzed using Image Lab 3.0 software.
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Beyotime Biotech, China). The purity and concentration of RNA were assessed using a Nano Photometer spectrophotometer (Thermo Fisher, USA). Subsequently, cDNA was synthesized from the RNA using a cDNA kit (BioRad). Real-time PCR was performed using qPCR Master Mix (Biotium), and the mRNA expression levels were analyzed using Bio-Rad CFX Manager software with GAPDH as the internal control. The primers used were provided by Sangon Biotech (Shanghai, China).
shRNA, siRNA, and plasmid interference
The shRNA, siRNAs, and plasmids were obtained from GenePharma (Shanghai, China) and lentiviral transfection was performed according to the manufacturer’s instructions. The transfection protocol and interference were previously described in our published study [4]. Relative gene and protein expression levels were used to assess the efficiency of the transfection.
Cell immunofluorescence (IF) staining
The medium was first removed from the 24-well plates. After three PBS washes, the cells were fixed with paraformaldehyde for 15 min before being promptly bilayed with 0.2% Triton X-100 for one hour. The cells were then inhibited for 30 min using serum. The coverslips were then treated with primary antibodies overnight at 4 °C. Following rinsing with PBS, the coverslips were incubated with secondary antibodies for 2 h in the dark. The coverslips were rinsed again with PBS before being stained with DAPI. The total number of positively stained cells within a view area of a fluorescence microscope was counted to measure the amount of protein expression.
Alkaline phosphatase (ALP) staining
After being planted in 12-well plates with 2.5 × 104 cells per well, BMSC was osteogenically induced for a week. The cells were subsequently fixed for 15 min in 4% paraformaldehyde. The wells were introduced to the BCIP/NBT working solution (300 µL) after three PBS washes. A one-hour incubation in the dark with the working solution was performed on the cells. Finally, using a laser microscope, the number of ALP-positive cells in each area was counted.
ALP activity assay
As directed by the manufacturer, the detection buffer, chromogenic substrate, and standard were fabricated. After lysing the cells from the various groups, the lysates were centrifuged at 3000 rpm for 15 min. For each sample well, blank well, and control well, the prepared reagents were added. A microplate reader was used to measure the OD value at 410 nm.
Alizarin red staining
2.5 ×104 BMSCs were planted in each well of 12-well plates, and they were then treated for 2 weeks in an osteogenic differentiation medium. A 15-minute fixation in 95% alcohol was then performed on the cells. The wells were put into an Alizarin Red solution (500 µL) after three PBS washes. The working solution was incubated with the cells for 15 min at room temperature. Then, ddH2O was three times circled each well to look for calcium nodules using a laser microscope. Finally, 10% cetylpyridinium chloride was used to dissolve the calcium nodules, and the OD value was measured at 570 nm.
Oil red O staining
BMSCs were seeded in 12-well plates at a density of 2.5 ×104 cells per well and incubated for two weeks in adipogenic differentiation medium. The medium was removed, and the cells were washed twice with PBS before being fixed for 20 min in 4% paraformaldehyde. The cells were washed 3 times with ddH2O before being incubated for five minutes in 60% isopropanol. The cells were then incubated for 10 min in an Oil Red O (Sigma, USA) solution. The cells were subsequently resuspended in 60% isopropanol and ddH2O. The tissue staining procedure was identical to that for cultured cells. A laser microscope observed and captured images.
TRAP staining of cells
Five days were spent incubating RAW264.7 cells in an osteoclast differentiation medium. The medium was eliminated, and we washed the cells three times in PBS before fixing them for 20 min in 4% paraformaldehyde and personalizing them for 5 min in 0.3% Triton X-100. As described previously [17], a leukocyte acid phosphatase staining kit (Sigma, USA) was then used to detect osteoclasts. Osteoclasts are TRAP-positive cells with more than three nuclei.
RNA sequencing
BMSCs were cultured in 6-well plates. We divided the cells into two groups and performed three replications for each group. One group was MAGL knockout cells, and the other group was transduced with a negative control shRNA. The two groups of cells were treated with DEX 1 µM for 2 days. Then the TRIzol reagent (Beyotime) was used to extract the total RNA. The final subsequent sequencing analysis of the samples was performed by OE Biotech (Shanghai, China). 150-bp paired-end reads were generated from the libraries using an Illumina platform. An alignment of the clean reads to Rattus norvegicus. Rnor 6.0 was then conducted. Cufflinks were used to calculate FPKM for each gene, and HTSeq-count was used to calculate read counts. To perform differential expression analysis, we used the R package DESeq (2012). We determined significant differential expression by setting the P value at 0.05 and the fold change at 2 or the fold change at 0.5. Gene expression patterns in different samples and groups were analyzed using hierarchical cluster analysis (HCA). DEGs were enriched using the hypergeometric distribution in GO and KEGG pathway enrichment analyses.
Measurement by liquid chromatography/mass spectrometry (LC-MS) of intracellular 2AG levels
BMSCs (1 ×105 cells/well) were cultured in 6-well plates and treated with MJN110 (0 µM, 1 µM, or 10 µM) in the presence and absence of DEX (1 µM) for 24 h. As internal standards, BMSCs were homogenized with CHCl3/CH3OH/Tris-HCl containing 10 pM of [2H]5-2AG. After that, drying, weighing, and pre-purification of the organic phase containing lipids were performed. The fractions obtained were then analyzed by LC/MS (Shimadzu, Kyoto, Japan). Analyses of 2AG were conducted using the selected mode of ion monitoring for LC/MS. We calculated 2AG levels based on the ratio of their area to the internal deuterated standard signal areas and normalized their amounts per mg of lipid extract to pM.
Animals and chemicals
The Laboratory Animal Center of Soochow University provided male Sprague Dawley (SD) rats (age: 10 weeks, weight: 400 ± 50 g). All rats (5 per cage) were housed in the specific pathogen-free (SPF) animal facility at Suzhou University Experimental Animal Center and were provided with standard feed and sterilized water. The rooms were maintained at constant temperatures and humidity levels, with 12-hour light cycles. As male rats typically have higher bone mass compared to female rats, in order to minimize gender-induced data bias, we exclusively used male rats in all of our experiments. No significant adverse events occurred in our study. As described in our previous study [4], the ONFH model induced by GC was established. Fifty SD rats were randomly assigned to one of five groups (n = 10): (1) Control group (DMSO only); (2) Model group (administration drugs: MP and LPS); (3) Treatment group (administration drugs: MP + LPS + MJN110); (4) AM251 + MJN110 group (administration drugs: MP + LPS + MJN110 + AM251); (5) AM630 + MJN110 group (administration drugs: MP + LPS + MJN110 + AM630). The DMSO, LPS, MP, MJN110, AM251, and AM630 doses utilized were based on those reported in earlier studies [18,19,20,21,22,23]. Six weeks after the establishment of the model, samples of the femoral head were obtained. All animal experiments were approved by Soochow University’s First Affiliated Hospital’s Ethics Committee.
Micro-CT scans
Using a SkyScan 1176, the femoral heads of the rats were scanned and analyzed (Bruker Micro-CT, Aartselaar, Belgium). Scan settings were as follows: 70 kV, 120 µA, and 200 ms at a voxel resolution of 12 μm in medium mode. For the bone parameter analysis of trabecular bone, the region of interest extends from 0.1 millimeters below the growth plate of the femoral head and is 1.5 mm long. DataViewer was used to acquire the axial section, sagittal section, and coronal section of each specimen. CT Analyzer software was utilized to evaluate the bone volume (BV), bone volume fraction (BV/TV), trabecular thickness (Tb. Th), and trabecular spacing (Tb. Sp).
H&E staining and TRAP staining
The femoral head samples were fixed with paraformaldehyde and decalcified in 10% EDTA. Then, the specimens were embedded and cut into 6-µm-thick sections. After H&E staining or TRAP staining, a laser microscope (Carl Zeiss, Oberkochen, Germany) was used to study the morphological changes by H&E staining and visualize the osteoclast density by TRAP staining. The detailed H&E staining and TRAP staining procedures were according to the previous study [24]. For the TRAP staining experiment, the cells containing purple staining and multiple nuclei were regarded as osteoclasts and manually counted. The number of osteoclasts within the view field was used to evaluate the level of bone resorption.
Immunohistochemistry (IHC) and tissue IF staining
In brief, after antigen retrieval with sodium citrate solution, the sections were stained overnight at 4 °C with RANKL primary antibody. The samples were then treated with the matching secondary antibodies. The positive staining was then developed using a DAB staining kit (Beyotime, China). The portions were then inspected using a laser microscope (Carl Zeiss, Germany). For tissue, IF staining, slices were blocked with BSA for 1 h and incubated with MAGL, OCN, and PPAR primary antibodies for 12 h. The slices were then washed with PBS and incubated for one hour with the matching secondary antibodies. DAPI was used to stain the nucleus. The tissue slices were then observed using a laser microscope (Carl Zeiss, Germany). The number of IHC-positive or IF-positive cells was quantified using Bioquant Osteo 2017.
Masson staining
After dewaxing and rehydrating the sections, they were stained with Weigert’s hematoxylin solution for 10 min. Afterward, they were separated with acidic alcohol and rinsed with ddH2O. Masson dye was then applied to the sections. Following washing with ddH2O, the working solution for the Ponceau stain was applied for 10 min. Washing the sections was carried out using a weak acid solution. Afterward, the sections were stained for 2 min with phospholipid acid solution, 1 min with a weak acid working solution, 2 min with aniline blue solution, and 1 min with a weak acid working solution. They were then dehydrated, hyalinized, and mounted using neutral resin.
Statistics
At least three times were conducted for each experiment. SPSS (version 20) was used for statistical analysis. The figures represent the average plus the standard deviation. Comparisons between two groups were performed using unpaired t-tests with Welch’s correction applied to the data. Using ANOVA, among multiple group differences were analyzed. Tukey’s test and Dunnett’s test were used to do the post hoc analysis. The significance level was determined by a P value of 0.05 or a P value of 0.01.

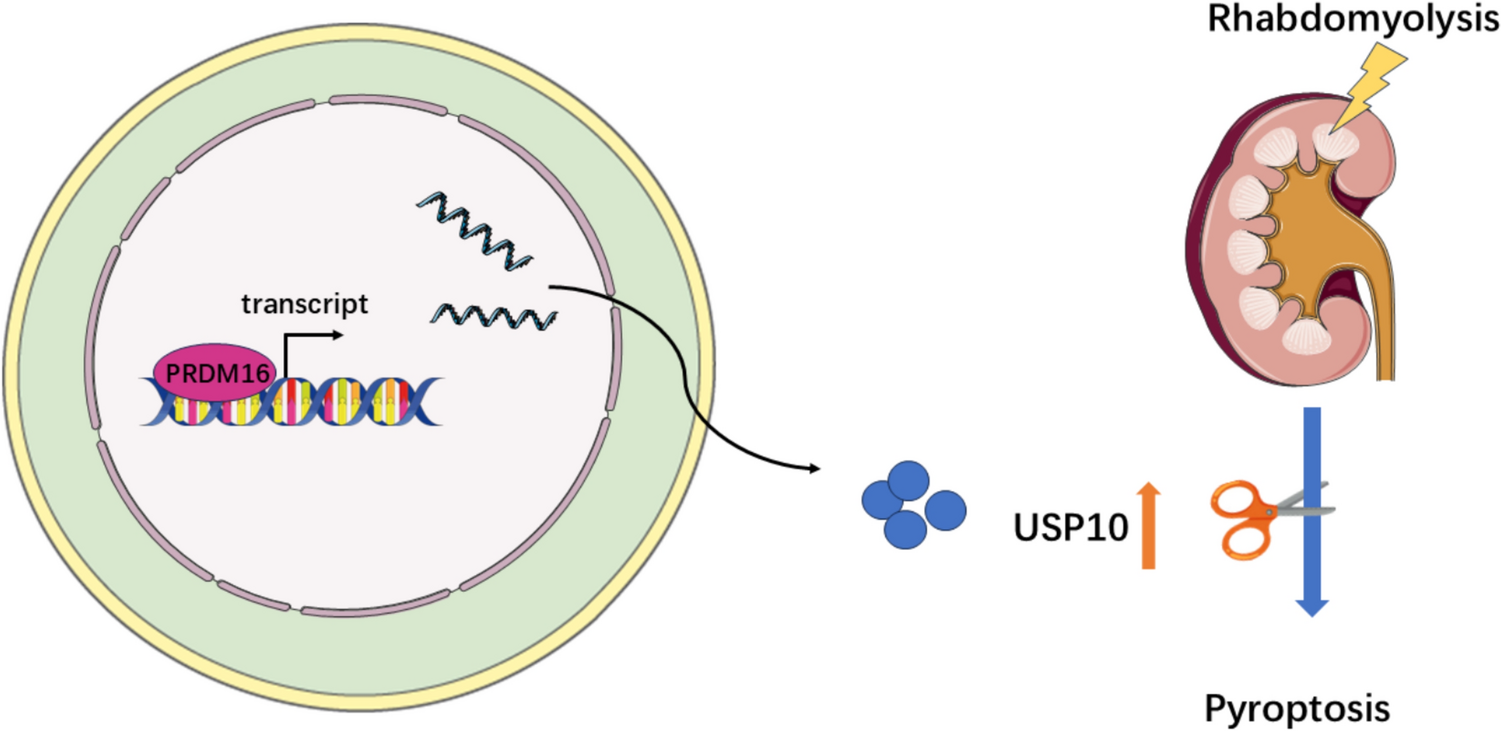


Comments (0)