
Remember me

A total of 396 strains of endophytic bacteria were isolated from different tissues of six medicinal plants, including Allium platyspathum Schrenk and Oxytropis merkensis Bge. The highest number of strains, 79, was isolated from Allium platyspathum Schrenk, followed by 77 strains from Oxytropis merkensis Bge (Fig. 1). Notably, strain MR4, isolated from Oxytropis merkensis Bge, exhibited significant biocontrol potential, inhibiting the cotton fusarium wilt by 60% and cotton verticillium wilt by 48.57%. This strain merits further investigation due to its strong biocontrol capabilities.
Fig. 1
Distribution of endophytic bacteria isolated from different plant tissues
Identification of strain MR4When inoculated onto LB medium using the three-zone streaking method and cultured at 37 °C, the colonies of strain MR4 were observed to be opaque and milky white in appearance, with a raised surface and wrinkled edges. When picked, the colonies exhibited a mucous consistency (Fig. 2). Strain MR4 was identified by amplifying partial segments of the 16S rDNA gene and the gyrA gene and then conducting similarity comparison analyses with data in GenBank. Based on the sequences of other standard spore-forming bacteria provided, ten strains with high similarity were selected to construct 16S phylogenetic trees and gyrA phylogenetic trees (Fig. 3A, B). The results indicated that strain MR4 clustered with Bacillus amyloliquefaciens, consistent with the annotation results in the NR database (Fig. 4). To further confirm this, nucleotide consistency analysis and heatmaps were generated by comparing MR4 with reference strains B. amyloliquefaciens FZB42, B. amyloliquefaciens DSM7, B. amyloliquefaciens B9601-Y2, and B. subtilis 168 (Fig. 5). The results showed that MR4 exhibited a similarity of over 97% with B. amyloliquefaciens FZB42 and B. amyloliquefaciens DSM7, while its similarity with B. subtilis 168 was below 95%, confirming MR4 as Bacillus amyloliquefaciens.
Fig. 2
MR4 endophytic bacteria colony diagram
Fig. 3
Identification of Bacillus amyloliquefaciens MR4 strain. Phylogenetic tree based on nucleotide sequence of 16S rRNA (A) and gyrA (B)
Fig. 4
Functional annotation results of NR database of Bacillus amyloliquefaciens MR4 genome
Fig. 5
Heatmap of nucleotide consistency between Bacillus amyloliquefaciens MR4 and four Bacillus strains
Antagonistic capability of strain MR4 against cotton verticillium and fusarium wiltsIn this study, we identified a bacterial strain, MR4, from the rhizosphere of medicinal plants that exhibited significant antagonistic effects against cotton verticillium and fusarium wilts. This finding aims to provide a scientific basis for enhancing disease resistance in cotton crops. In plate co-cultivation experiments, both the supernatant and cell suspension of the MR4 strain demonstrated effective inhibitory effects against cotton verticillium wilt and fusarium wilt (Fig. 6). Specifically, the sterile fermentation supernatant of MR4 inhibited cotton verticillium wilt by 46.54% (Table 2A) and fusarium wilt by 58.09% (Table 2C). Similarly, the MR4 bacterial suspension inhibited cotton verticillium wilt by 58.8% (Table 2B) and fusarium wilt by 72.02% (Table 2D). By quantitatively assessing the inhibitory effects of endophytic bacteria on pathogen growth, this study provides insights for further exploring the potential antagonistic mechanisms of endophytic bacteria and highlights the potential of selecting effective antagonistic bacteria from plant rhizosphere environments as biocontrol agents (Tyśkiewicz et al. 2022).
Fig. 6
Image of the inhibitory effect of sterile fermentation supernatant (15%) and bacterial suspension on fusarium wilt and verticillium wilt pathogens in cotton. (Note: A, B the inhibitory effects of MR4 strain supernatant and bacterial suspension on cotton fusarium wilt, respectively. C, D The inhibitory effects of MR4 non-fermentation supernatant and bacterial suspension on verticillium wilt of cotton, respectively)
Table 2 Inhibition rate of (A) endophyte aseptic fermentation supernatant on cotton fusarium wilt, (B) endophytic suspensions on cotton fusarium wilt, (C) endophytic supernatant of aseptic fermentation on verticillium wilt of cotton, and (D) endophytic suspensions against verticillium wilt of cottonAntimicrobial activity genes in MR4Using 15 pairs of primers, PCR amplification of the genes responsible for the synthesis of antimicrobial substances in the antagonistic strain MR4 was performed (Fig. 7). The results showed that MR4 could amplify fragments of the genes srfAA, fenB, ituA, ituC, bmyA, bacAB, bacD, bacA, baseS, dhbA, ituD, and mycB, with approximate sizes of 1600 bp, 1600 bp, 1047 bp, 594 bp, 1200 bp, 500 bp, 815 bp, 1200 bp, 1550 bp, 1350 bp, 1203 bp, and 2000 bp, respectively. These genes encode for seven different non-ribosomal peptides: surfactin, fengycin, iturin, bacillomycin, bacilysin, iturin again, and mycosubtilin. These non-ribosomal peptides exhibit a broad spectrum of antimicrobial activities. For instance, fengycin effectively inhibits fungal diseases in plant root systems, while surfactin demonstrates exceptional performance in promoting plant growth with two polyketide compounds (bacillaene and bacillibactin). Bacillaene inhibits bacterial growth by interfering with the cell division process of pathogenic bacteria (Nakayinga et al. 2021). Bacillibactin, an iron-chelating siderophore, facilitates bacterial iron uptake by binding to iron ions in the environment, thus providing a survival advantage under iron-limited conditions. Although its direct antimicrobial activity may not be pronounced, it plays a crucial role in bacterial growth and survival, indirectly affecting interactions with plants (Luo et al. 2022). The gene for the synthesis of difficidin (dfnA) was not amplified. These results indicate that strain MR4 possesses the capability to synthesize at least eight different types of antimicrobial substances, and it has excellent potential of biocontrol and growth promotion.
Fig. 7
PCR results of MR4 gene encoding antimicrobial substance
Genome features of MR4The genomic DNA of MR4, extracted and analyzed by agarose gel electrophoresis, displayed a clear band, indicating that the extracted DNA met the quality standards (Fig. 8). Through whole-genome sequencing analysis, we determined the total genome size of the antagonistic strain MR4 to be 4,017,872 base pairs (bp), with a total of 4191 genes encoded. These encoded genes occupy 89.9% of the entire genome. The total length of the encoded genes reaches 3,611,934 bp, with an average gene length of 862 bp and a GC content of 47.14%. The genome consists of a single chromosome and one plasmid. Specifically, the length of the chromosome is 3,937,108 bp with a GC content of 46.58%, while the plasmid length is 80,764 bp with a GC content of 38% (Fig. 9A, B). Within the genome of MR4, we identified 87 tRNA genes, 6 sRNA genes, and 9 each of 5S rRNA, 16S rRNA, and 23S rRNA genes. In addition, eight genomic islands, ten potential prophages, and three CRISPR sequences were discovered. In the gene function annotation process, we identified 3391, 3013, 2748, 3952, and 2748 genes in the Swiss-Prot, COG, GO, KEGG, and Pfam databases, respectively. These annotations provide crucial information about the functional characteristics of the MR4 genome (Table 3). These whole-genome analysis results lay the foundation for further exploration of the biological functions of MR4.
Fig. 8
Electrophoretic profile of MR4 (Note: Number 3 refers to MR4)
Fig. 9
MR4 genome circle map (A) and plasmid profile (B). (Note: A The outermost circle is the position coordinates of the genome sequence. From the outside to the inside, it is the result of gene function annotation, ncRNA, genome GC content, and genome GC skew value. B From the outside to the inside, the pictures are COG functional annotation classification genes, genomic sequence position coordinates, genomic GC content, and genomic GC skew value)
Table 3 MR4 annotation resultsGene function annotation analysisBasic function annotation analysisThe GO database is subdivided into three main categories: cellular component, molecular function, and biological process. In the cellular component category, 497 genes are related to cells, with 409 genes associated with the cell membrane. In the molecular function category, the most common annotations are catalytic activity and binding functions, linked to 1575 and 1311 genes, respectively. In the biological process category, the highest number of genes is related to metabolism, totaling 1613 genes (Fig. 10). This suggests that the MR4 strain may possess strong metabolic capabilities and environmental adaptability, which could be crucial for its survival in the environment and interactions with hosts. Protein annotation of MR4 was performed using the COG database. The classification of the 3390 genes annotated by COG for strain MR4 is shown in Fig. 11. Among them, there are 1015 genes related to amino acid transport and metabolism. Additionally, the most abundant annotation is for transcription, with 295 genes, accounting for 8.7% of the annotated genes. This is followed by genes related to carbohydrate transport and metabolism, as well as general function prediction, with 247 and 246 genes, respectively (Fig. 11). In the KEGG database, the MR4 genome has been annotated with 3764 genes, covering six major categories, including cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and biological systems. Particularly noteworthy are the metabolic pathways and environmental information processing pathways, encompassing 1720 and 286 genes (Fig. 12). Studying these pathways and genes can provide deeper insights into the microbial ability to degrade harmful substances and adapt to the environment. Interpreting this information will provide important clues for understanding the biological characteristics and potential applications of microorganisms. In the Pfam (Protein families) database, the most common functional domain is the P-loop containing the NTPase superfamily, with 1220 genes containing this structural domain. The CAZy (Carbohydrate-Active enZYmes) database identified 166 genes. The Swiss-Prot database indicates the presence of three ATP-binding proteins LnrL (Linearmycin Resistance ATP-binding Protein LnrL) with linearmycin resistance activity, as well as three polyketide synthases PksM (Polyketide Synthase PksM). The ATP-binding protein LnrL conferring linearmycin resistance is an ABC transporter protein, primarily aiding bacterial cells in excluding harmful substances and speculated to play a crucial role in biocontrol. However, the specific mechanism of action of LnrL protein in cell signaling and disease progression remains unclear and requires further investigation (Zhang et al. 2023). Polyketide synthase PksM is involved in the synthesis of polyketide compounds, which often possess significant biological activities, including antibiotic effects. The comprehensive annotation results from multiple databases indicate that Bacillus amyloliquefaciens has strong biocontrol potential and merits further exploration.
Fig. 10
Functional annotation results of GO database of Bacillus amyloliquefaciens MR4 genome
Fig. 11
Functional annotation results of COG database of Bacillus amyloliquefaciens MR4 genome
Fig. 12
Functional annotation results of KEGG database of Bacillus amyloliquefaciens MR4 genome
Prediction of MR4 secondary metabolite gene clustersWe utilized the antiSMASH database for prediction and identified a total of 13 secondary metabolite biosynthetic gene clusters in the MR4 genome (Table 4, Fig. 13). These clusters include NRPS, betalactone, transAT-PKS, lanthipeptide-class-ii, NRPS, T3PKS, NRPS, T3PKS, transAT-PKS, PKS-like, NRPS, RiPP-like other, LAPthiopeptide, terpene, and transAT-PKS. Among them, trans-PKS and terpene are dominant gene clusters, each composed of two gene clusters. The transAT-PKS gene cluster may play a role in the production of specific polyketide secondary metabolites, such as antibiotics, pigments, or toxins. The identified two terpene gene clusters are crucial components for the biosynthesis of terpene compounds in plants, playing a significant role in plant biosynthesis and the interaction between plants and the environment (Chu et al. 2011). We found that the majority of the predicted gene clusters are associated with antifungal effects.
Table 4 MR4 secondary metabolic gene clusterFig. 13
Annotation results of the secondary metabolite gene clusters in Bacillus amyloliquefaciens MR4
Antagonistic potentialThe Pathogen-Host Interaction Database (PHI) is an important biological information resource with significant implications for studying potential target genes in drug intervention. The PHI database records various gene variations related to pathogen traits. The annotation results of MR4 in the PHI database (Fig. 14) indicate that gene mutations that weaken pathogen virulence are the most common, with a total of 203 mutations. Next are gene mutations that do not affect pathogenicity, totaling 73. Additionally, there are relatively more gene mutations that enhance pathogen virulence, reaching 29. Gene mutations that completely lose pathogenicity amount to 9, while lethal gene mutations total 13. These data not only reveal the number of mutated genes but also indicate their specific locations in the genome, providing a solid theoretical basis and direction for further in-depth studies of this strain. The Virulence Factor Database (VFDB) focuses on studying the pathogenic factors of pathogenic bacteria, chlamydia, and other pathogenic microorganisms. It provides species information on virulence genes, basic characteristics descriptions, and detailed explanations of virulence gene functions and pathogenic mechanisms. Through VFDB annotation analysis, 198 gene annotations were obtained, mainly involving bacterial adhesion, secretion system proteins, and transcription factors. In the results from the Antibiotic Resistance Genes Database (ARDB), annotations for seven types of resistance genes were identified, including lincomycin, phosphomycin, tetracycline, and others. It is speculated that Bacillus amyloliquefaciens may express resistance to these antibiotics and pump them out through efflux resistance proteins to protect itself and develop resistance.
Fig. 14
Functional annotation results of PHI database of Bacillus amyloliquefaciens MR4 genome
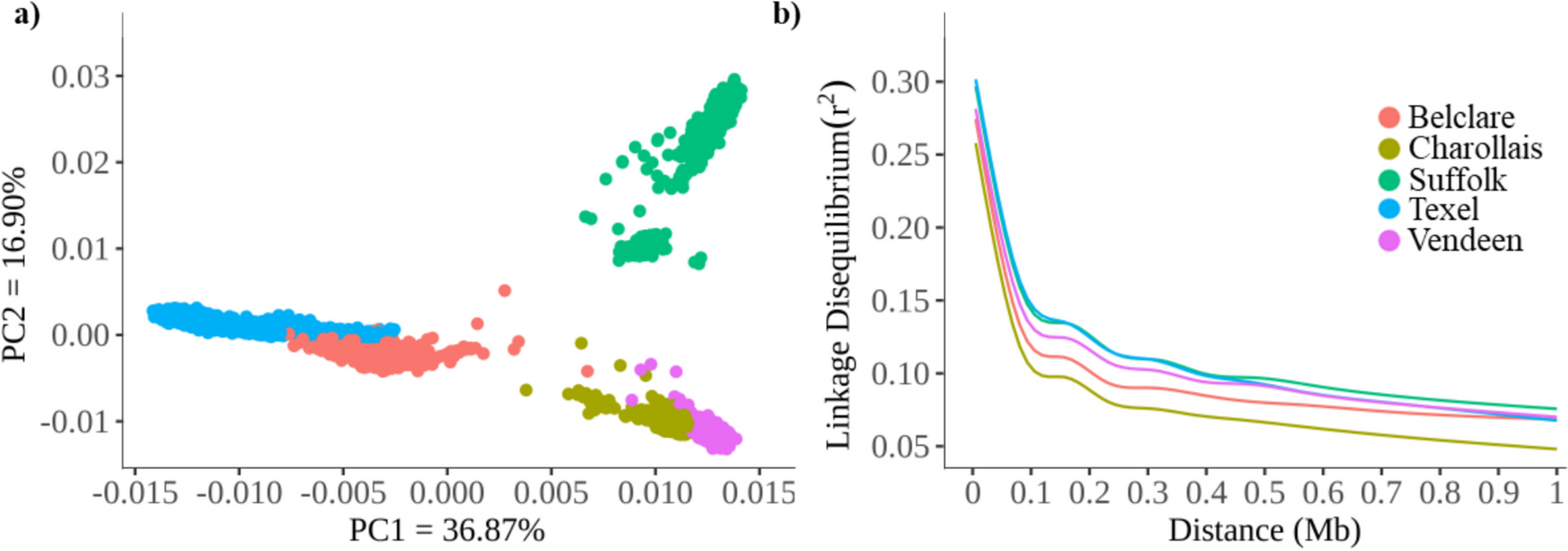
Comparison of MR4 with Bacillus spp. strainsWe found that the size of the MR4 genome is similar to that of four Bacillus strains, with the closest match being to B. amyloliquefaciens DSM7. The GC content is most similar to that of B. amyloliquefaciens FZB42, followed by B. amyloliquefaciens DSM7. The number of tRNAs is comparable to that of B. subtilis 168 and lower than the other three strains of starch-degrading Bacillus (Table 5). The genome of this strain exhibits widespread evolutionary events such as gene inversion and gene recombination. By comparing homologous regions in the genome sequences using alignment tools, evolutionary processes can be determined. To assess the evolutionary distance between the five strains, their whole-genome sequences were compared using the MAUVE program with default parameters (Fig. 15). The alignment results show that there are significant gene insertions or deletions and locally collinear blocks (LCBs) inversions between DSM7, 168, and B9601-Y2. The genomes of strains MR4 and FZB42 exhibit good collinearity, with no extensive rearrangements or deletions, but transpositions and insertions are still present. Overall, strain MR4 shows a closer genetic relationship with B. amyloliquefaciens FZB42 among the five strains of Bacillus.
Table 5 Basic characteristics of the whole-genome sequences of MR4 and four related strainsFig. 15
Collinearity relationship diagram between Bacillus amyloliquefaciens MR4 and four model strains
Analysis of the biocontrol and growth-promoting potential of MR4Through the pot culture experiment, we observed that the two cotton varieties, Xin Hai 21 and Xin Lu Zhong 63, demonstrated better growth and healthier conditions after inoculation with the MR4 strain compared to the untreated control group. Specifically, the cotton plants inoculated with MR4 had broader and fuller leaves with a more vibrant green color, whereas the untreated control group exhibited slight leaf chlorosis and narrower leaves (as shown in Fig. 16).
Fig. 16
A Xin Hai 21 control; B Xin Hai 21 treated with MR4; C Xin Lu Zhong 63 control; D Xin Lu Zhong 63 treated with MR4
According to the agronomic trait measurements, the average plant height of Xin Hai 21 cotton inoculated with MR4 was 32.4 cm, the average fresh weight was 1.8 g, the average dry weight was 0.2 g, and the average leaf count was 5 leaves. In contrast, the corresponding data for the Xin Hai 21 control group were 25.7 cm, 1.6 g, 0.17 g, and 4 leaves, respectively. For Xin Lu Zhong 63 cotton, the average plant height after MR4 inoculation was 31.0 cm, the average fresh weight was 1.6 g, the average dry weight was 0.18 g, and the leaf count was 5 leaves. The corresponding data for the control group were 28.3 cm, 1.5 g, 0.17 g, and 5 leaves (detailed in Supplementary Tables 1 and 2).
Overall, the cotton plants inoculated with MR4 showed superior performance in terms of average plant height, fresh weight, dry weight, and leaf count compared to the control group. This indicates that the MR4 strain has the potential to promote cotton growth and enhance plant health. These results support the potential application of the MR4 strain as a biocontrol agent and growth promoter.
Comments (0)