
Remember me



The first new patient is a boy, now aged 9. He is the second child of healthy nonconsanguineous parents. He was born by vaginal delivery after an uneventful pregnancy. Growth parameters at birth were normal (W 3120 g, L 49 cm, HC 33 cm) and he experienced no perinatal distress (Apgar 10/10). Multiple angiomas (face, nape of neck, left buttock) were found, so that abdominal, medullary and brain US were performed, without particular findings.
At 3 weeks of age, the child was taken to the ER for episodes of projectile vomiting: abdominal US excluded a pyloric stenosis, but bilateral midriasis and hypotonia were found, so that the child underwent further exams. An ophthalmic checkup confirmed pupillary midriasis, with mild bilateral iris hypoplasia, hypopigmented fundus, and the presence of epipupillary remnants of Wackendorf's membrane. Brain MRI was normal, as well as a cardiological examination with echocardiogram.
Because of the presence of delay in early motor development, he started physiotherapy and was later brought to our attention at the age of 11 months for further consultation.
He was in good general health, except for a slight underweight (L 71 cm = -1.31 SD, W 7.4 kg = -2.7 SD, HC 45 cm = -0.86 SD). He presented flat angiomas at forehead, both upper eyelids, upper lip, and left buttock. Pupils were mydriatic, with torpid light reflex; visual pursuit was discontinuous. Diffuse muscle hypotonia was present and the child was able to control the head but not to sit autonomously. Brain MRI was repeated and confirmed to be normal; visual evoked potentials (VEP) and electroretinogram (ERG) showed the presence of scotopic-dominant retinal alterations, with photopic responses and visual responses in the normal range; brainstem auditory evoked potentials (BAEP) and electroencephalogram (EEG) were normal.
Despite the absence of cerebellar atrophy, Gillespie syndrome was suspected and confirmed by ITPR1 genetic analysis, which identified a heterozygous de novo pathogenic variant [ITPR1(NM_001378452.1):c.7660G > A p.(Gly2554Arg)], affecting residue Gly2554 that represents a mutational hotspot for Gillespie syndrome [5].
The child was followed with annual clinical controls. He showed a constant improvement of psychomotor development: as for motor skills, he became able to sit independently by the age of 2 and to walk with help at the age of 4, while language development progression was initially slow but reached normal levels during school-age.
Growing up, he developed an ataxic syndrome, characterized by oculomotor dyspraxia, dysarthria, dysmetria, tremor, truncal titubation, and gait instability. SARA score at 8 years old was 23 (Gait 5, Stance 4, Sitting 2, Speech disturbance 4, Finger chase 2, Nose-finger test 2, Fast alternating hands movements 2, Heel-shin slide 2).
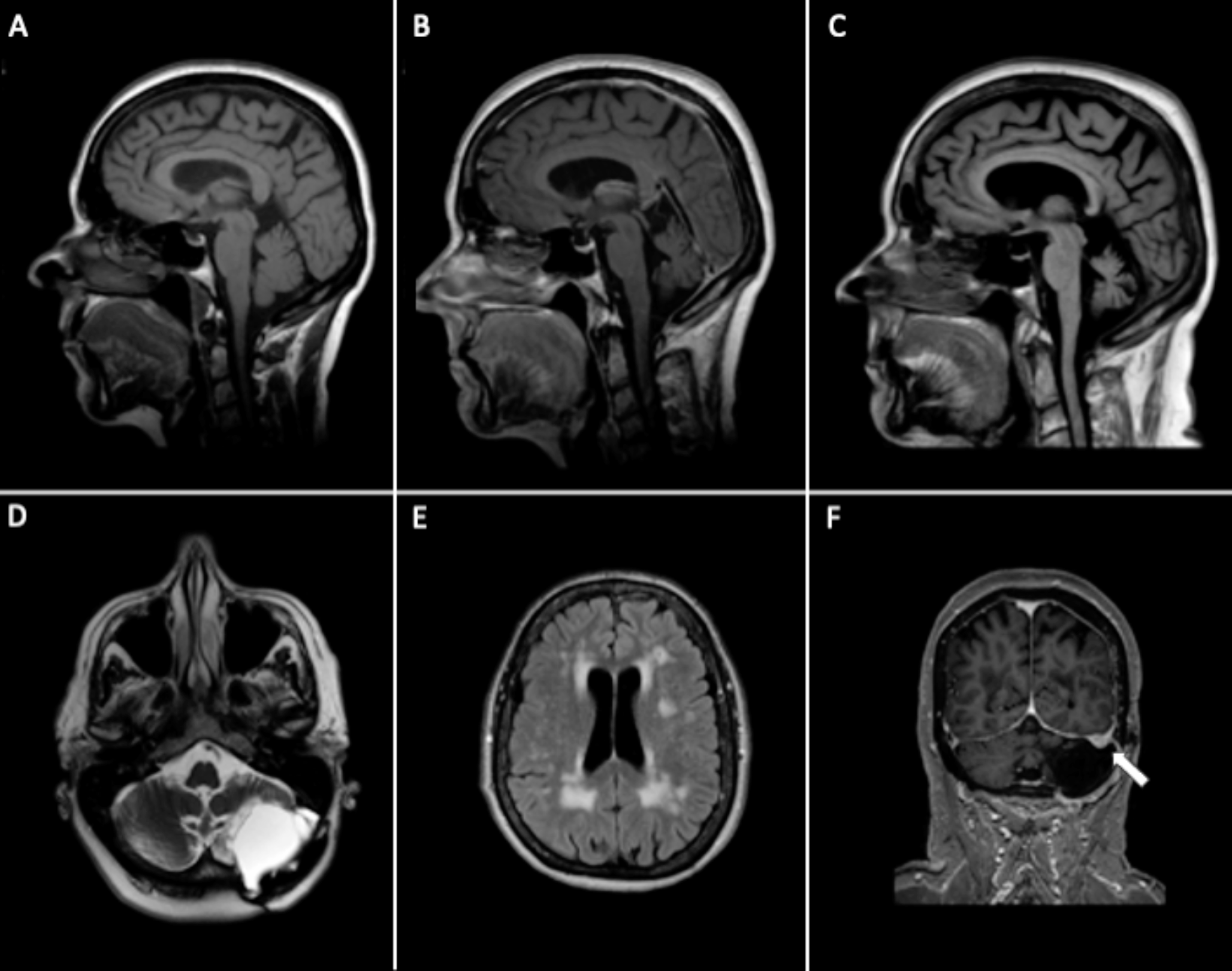
A third brain MRI performed at the age of 4 years and 2 months documented an involutional enlargement of the cerebellar-vermian cerebrospinal fluid spaces, with cerebellar vermis and hemispheres atrophy, hyperintense signal alterations of cerebellar gyrus and superior cerebellar peduncles in FLAIR images and shaded hyperintense signal alterations in T2/FLAIR images of occipital subcortical white matter (Fig. 1). A subsequent MRI control at 8 years appeared unchanged.
Fig. 1
Brain MRI of Case 1 (Patient 6). Panel 1—Brain MRI at the age of 4 months: Sagittal TSE-T1w (1a) and coronal TSE-T2w (1b) showed no significant morphological abnormality, with asymptomatic small pineal cyst; axial T2w-FLAIR (1c) revealed a mild delay in deep white matter myelination. Panel 2—Follow-up MRI at the age of 4 years: Sagittal TSE-T1w and coronal TSE-T2w showed marked cerebellar and vermis atrophy (2a, arrow) with folia prominence of the superior aspect of cerebellar hemispheres (2b, star). Residual altered signal intensity on T2w was still visible within deep white matter of occipital lobes (2c, dotted)
BAEP, PEV, ERG were also repeated twice, resulting normal. EEG registration at 5 and 8 years showed the presence of multifocal epileptic anomalies in sleep, but the child never reported seizures.
Last cognitive evaluation at the age of 8 confirmed cognitive skills within normal limits in all the considered domains, except for Processing Speed score, influenced by severe deficits in visual-perceptual analysis and visuomotor integration. The overall IQ score at Wechsler Intelligence Scale for Children (WISC-IV) was 70 (subscales: Verbal Comprehension 74, Perceptual Reasoning 82, Working Memory 94, Processing Speed 59). The evaluation also evidenced difficulties in school learning processes and some sub-clinical behavioral problems in attention, and emotional and social responses.
Case 2 (Patient 19)The second new case is a girl, now aged 17 years. She is the first child of healthy nonconsanguineous parents. She was brought to our attention at the age of 2 for the occurrence of developmental delay and a peculiar iris abnormality, initially diagnosed as a partial coloboma with a suspect of glaucoma but later characterized as bilateral iris hypoplasia.
Neurological examination was suggestive for an ataxic syndrome, with ocular motility anomaly, trunk titubation and instability with difficulties in reaching and maintaining the stance position.
The first brain MRI, performed at the age of 2, revealed global cerebellar atrophy with predominant involvement of cerebellar vermis, and a slight T2-hyperintensity of cerebellar cortex. The patient underwent genetic tests, including array-CGH and sequencing of FOXC1, PITX2, and PAX6 genes, all resulting normal. There was a suspicion of Gillespie syndrome, but the genetic bases of the disease were not yet known at that time.
The girl was followed over the years. The administration of Griffiths Scales for Mental Development (GSMD) at 2 years revealed a moderate global developmental delay (DQ 44) with significant weaknesses in motor skills. At the age of 5 she became able to stand independently and walk few steps with help or with a walking frame. Clinical presentation has been stable during follow-up until the age of 17 years, without any worsening, and is currently characterized by an overt ataxic syndrome with oculomotor and orobuccal dyspraxia, dysarthria, truncal titubation in sitting and standing position, dysmetria, intention tremor exaggerated from anxiety or stress, and ataxic gait with severe instability in half turnings. Mild global hypotonia is present, but with vivid deep tendon reflexes and a slight bilateral Babinski sign. At the age of 16, she had an overall SARA score of 25 (Gait 7, Stance 4, Sitting 1, Speech disturbance 4, Finger chase 2, Nose-finger test 3, Fast alternating hand movement 2, Heel-shin slide 2). Consistently with iris hypoplasia, she shows bilateral mydriasis and a very slow fotomotor reflex. The global developmental delay evolved into moderate intellectual disability (no standardized intelligence tests available).
Despite a stable clinical presentation, an evolution of cerebellar atrophy was observed at the age of 3, but later examinations (at 4 and 5 years of age) showed no further progression.
Ophthalmologic and orthoptist controls documented an overall improvement with normal intraocular pressure. VEP and ERG showed a mild alteration of retino-cortical transmission and cortical activation; EEGs never disclosed significant anomalies.
ITPR testing revealed a heterozygous de novo variant: ITPR1(NM_001378452.1):c.7661G > T p.(Gly2554Val). The variant was never reported or annotated before (absent from GnomAD with an 88.42% coverage) but involves the same mutational hotspot residue (Gly2554) as in Patient 1. Including de novo criteria, it is classifiable as pathogenic according to ACMG guidelines (criteria PM2, PM5, PP2, PS2).
Comments (0)