Experimental design
The manuscript for this animal study was prepared following the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Seven-week-old male C57BL/6J apoE−/−mice (Cyagen Biotechnology Co., Ltd., Suzhou, Jiangsu, China) were raised in an experimental animal center in a specific pathogen-free environment with a 12-h light/dark cycle and free access to water and food and were fed an HFD after one week of adaptive feeding. All procedures were carried out according to the protocol approved by the Animal Care and Use Committee (protocol number: 2020-0041). The sample size was determined by G*power 3 (https://stats.idre.ucla.edu/other/gpower/, UCLA, USA), which referenced data from previous experiments.12 In this study, to achieve P < 0.05 and 95% power, n = 17 per group was needed. Considering the extended period, differences in diets, and the recommendations of the American Heart Association,50 the final decision on sample size was n = 19 in each group. Thirty-eight mice were randomly assigned to two groups (n = 19 per group) using random numbers generated by a computer. After one week of acclimatization, CAP was induced in the CAP group by P. gingivalis infection in the first and second molars of the bilateral maxillary region under anesthesia (0.1 mL per 10 g 1% pentobarbital; Sigma‒Aldrich, Shanghai, China). After the pulp chamber was exposed, a small piece of sterile cotton containing 0.2 µL of 108 colony-forming units (CFU)/mL P. gingivalis (logarithmic growth phase, strain ATCC33277) was verified as ATCC33277 by 16S full-length sequencing against the NCBI database. Supplementary 6 a-b) was immediately inserted into the pulp cavity, followed by immediate temporary sealing with zinc oxide. The Con group was anesthetized only. All mice were fed an HFD to induce atherosclerosis. The specific operation schedule of the experiment is shown in the time flow diagram (Supplementary Data 1c).
Staining and quantification of atherosclerotic lesions in the aortic arch
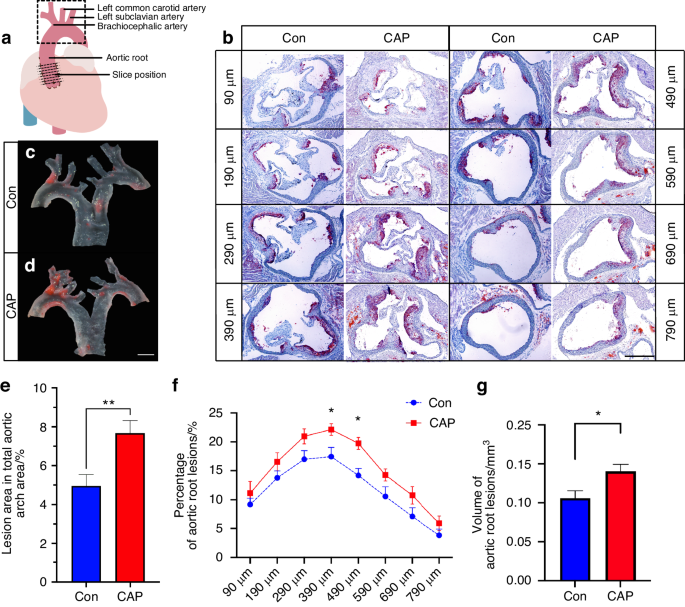
The aortic arch was carefully dissected under a stereomicroscope (Leica Camera GmbH, Solms, Germany) and fixed in 4% paraformaldehyde for 24 h. The aortic arch was then opened along the lateral and medial curvatures. As described previously, the lesions were stained with Oil Red O (Sigma‒Aldrich, Shanghai, China).51 The images were obtained by stereomicroscopy and analyzed with Fiji (NIH, Bethesda, MD, USA; https://imagej.net/downloads). Considering individual differences in arterial plaque as well as the aortic arch, the percentage of atherosclerotic lesions was determined by dividing the area of red area plaques stained by Oil Red O by the area of the overall aortic arch after microdissection. One sample in the Con group could not be used to determine the extent of lesions in the aortic arch because of errors during microdissection.
Staining and quantification of atherosclerotic lesions in the aortic root
The evaluation of atherosclerotic lesions in the aortic root was performed mainly with reference to the recommended methodology.51 Cardiac tissues were removed from 4% paraformaldehyde solution, routinely dehydrated in a sucrose gradient (10% sucrose, 20% sucrose, 30% sucrose), embedded in OCT, and sectioned in a freezer to a thickness of 10 μm by a frozen slicer (NX70, Thermo Fisher Scientific, Sweden). The sections were retained starting at the point where three intact valves were observed at the root of the aorta51 (eight sections 90–790 μm from the aortic sinus were collected) and stored at −20 °C for Oil Red O staining. The frozen sections were rewarmed at room temperature and washed in distilled water to remove the embedding agent. The sections were washed with 60% isopropyl alcohol, stained with Oil Red O working solution, stained with 60% isopropyl alcohol under a microscope, and washed with water immediately. Glycerol gelatin was used to seal the sections. Oil Red O staining of the aortic root was used to evaluate the extent of atherosclerosis. Images were taken under an inverted microscope, and the results were measured and analyzed using ImageJ. Eight levels of atherosclerotic lesion area were outlined by ImageJ, and the atherosclerotic volume of each sample was calculated by calculating the area under the curve of the 8-level atherosclerotic area line graph band edges.
Micro-CT analysis
The paraformaldehyde-fixed jawbone samples were placed on a micro-CT system (μCT100, Seanco Medical, Bassersdorf, Switzerland) and scanned along the long axis of the specimen to obtain a continuous micro-CT image with a resolution of 1 024 × 1 024, a pixel size of 15 μm × 15 μm, and a layer spacing of 15 μm. The periapical bone resorption area was reconstructed using Mimics software.
16S rRNA sequencing of the gut microbiota
Fresh fecal samples were collected at 19 weeks of age, and the samples were subsequently transferred to dry ice and stored at −80 °C. Microbial DNA was extracted using an EZNA® kit (Omega Bio-Tek, Norcross, GA, USA). A PCR thermocycler system (GeneAmp 9700, ABI, USA) was used to amplify the V3–V4 variable region (primers 338F and 806R), after which the products were extracted from 2% agarose gels using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union, CA, USA). Further purification and quantification were conducted using QuantiFluor™-ST (Promega, USA) according to the manufacturer’s protocol. The GREENGENES database was used for microbiota annotation. LEfSe was used to identify bacteria that differed in abundance between the samples.52 The core diversity plug-in of QIIME2 was used to calculate diversity metrics and the alpha diversity index. The Bray‒Curtis distance was used as an index to explore beta diversity, which was determined via PCoA.53
Fecal microbiota transplantation
Two FMT groups were established: the CAP recipient group (FMT group receiving CAP, FMT-CAP group, n = 13) and the control recipient group (FMT group receiving Con, FMT-Con group, n = 13). The recipient mice in both groups were pretreated with a mixture of four antibiotics (ampicillin 1 g/L, neomycin 0.5 g/L, vancomycin 0.5 g/L, and metronidazole 1 g/L) for 3 days,20 and the feces of the mice in the donor group were collected to prepare the bacterial suspension, which was administered to the recipient group by gavage. In the first week, the frequency of gavage was maintained at two times a week, and then the dosage was changed to one once a week for 12 consecutive weeks. Fresh feces (200 mg) were collected from the donor mice, and 4 mL of sterilized saline was added. After mixing, the supernatant was left to stand, and the recipient mice were administered the supernatant by gavage.
Metabolomics analysis
Mouse feces (100 mg) were ground separately in liquid nitrogen and then resuspended by vortexing with precooled 80% methanol. The samples were incubated on ice for 5 min and then centrifuged at 15 000×g for 20 min at 4 °C. Some of the supernatants were diluted to a final concentration of 53% methanol with LC‒MS-grade water. The sample was then transferred to a fresh Eppendorf tube and centrifuged at 15 000×g for 20 min at 4 °C. Finally, the supernatant was injected into the LC‒MS system for analysis.54 LC‒MS analysis was performed using a Vanquish UHPLC system (Thermo Fisher, Germany) and an Orbitrap Q ExactiveTM HF mass spectrometer (Thermo Fisher, Germany). The samples were injected onto a Hypesil Gold column (100 × 2.1 mm2, 1.9 μm) using a 17-min linear gradient at a flow rate of 0.2 mL/min. The eluents for the positive polarity mode were eluent A (0.1% FA, water) and eluent B (methanol). The negative polar mode eluents used were eluent A (5 mM ammonium acetate, pH 9.0) and eluent B (methanol). The Q ExactiveTM HF mass spectrometer was operated in positive/negative polar mode with a spray voltage of 3.2 kV, a capillary temperature of 320 °C, an intrathecal gas flow rate of 40 arb and an auxiliary gas flow rate of 10 arb. The raw data files were processed using Compound Discoverer 3.1 (CD3.1; Thermo Fisher, Germany) for peak matching, peak extraction, and quantification of each metabolite. The peak intensities were normalized to the total spectral intensity. The normalized data were used to predict molecular formulae based on additional ions, molecular ion peaks, and fragment ions. The peaks were subsequently matched against the mzCloud (https://www.mzcloud.org/), mzVault, and MassList databases to obtain accurate qualitative and relative quantitative results. The KEGG database (https://www.genome.jp/kegg/pathway.html), HMDB (https://hmdb.ca/metabolites), and LIPIDMaps database (http://www.lipidmaps.org/) were used for metabolite annotation. The R package MetaboAnalystR55 was used for data normalization and OPLS-DA. To bring the data closer to the normal parameters, MedianNorm, LogNorm, and AutoNorm were used. We applied univariate analysis of variance (t-test) to calculate the statistical significance (P-value).
Fluorescein isothiocyanate-dextran 4 (FD-4) assay for intestinal permeability
Intestinal permeability was measured by measuring FD-4 (60842-46-8, Sigma‒Aldrich, Shanghai, China) in 17-week-old mice. FD-4 was dissolved in saline solution and administered to mice by gavage at a concentration of 22 mg/mL in a 0.5 mL volume. After 5 h of gavage, the eyeballs were removed to collect blood, which was centrifuged at 10 000 r/min for 10 min at 4 °C. A total of 50 μL of the plasma was diluted in an equal amount of PBS (pH 7.4), and the concentration of FD-4 was determined by fluorescence spectroscopy using an excitation wavelength of 485 nm as a standard for serial dilutions and an emission wavelength of 528 nm. 7.4), and the concentration of FD-4 was determined using fluorescence spectroscopy at an excitation wavelength of 485 nm and an emission wavelength of 528 nm, with serially diluted samples used as standards. All samples and standards were measured three times.
AB-PAS staining
The experiments were performed according to the instructions of the AB-PAS kit (G1285, Beijing, Solarbio Technology Co.). Sections were stained in 3% acetic acid solution with 1% Alcian blue solution (pH 2.5) for 15–20 min. The sections were then rinsed in deionized water for 2 minutes and oxidized in 0.5% periodate solution for 5 min. The slides were rinsed and stained with Schiff’s reagent for 15 min, followed by hematoxylin staining.
Immunofluorescence staining to evaluate the mechanical and mucus barrier functions of the intestine
Paraffin-embedded tissue sections were deparaffinized and rehydrated, and citric acid was used to retrieve antigens at high pressure and temperature. After the goat sera were collected, they were incubated with primary antibodies (ZO-1, 1:50; 61-7300, Thermo Fisher Scientific, USA; Occludin, 1:50; 71-1500, Thermo Fisher Scientific, USA; Claudin, 1:50; Ab211737, Abcam, USA; and MUC-2, 1:50; Ab272692, Abcam, USA) overnight at 4 °C. A goat anti-rabbit fluorescent secondary antibody (1:1 000; Affinity Biosciences, S0008) was used. DAPI was used to visualize the b nuclei. Pictures were taken using an Olympus microscope (Olympus, Tokyo, Japan).
Western blot analysis
Twenty grams of mouse intestinal tissue was weighed, washed with cold PBS 2–3 times, and cut into small pieces. Then, 250 μL of RIPA lysis solution was added, and the sample was ground in a tissue-grinding apparatus. The homogenate was transferred to a centrifuge tube, shaken, and centrifuged at 12 000×g for 5 min, after which the supernatant was collected as the total protein solution. A BCA protein assay kit (Beyotime Biotechnology, Shanghai) was used to quantify the extracted proteins. After separation by SDS‒PAGE, the proteins were transferred to PVDF membranes. The PVDF membrane was blocked with 5% skim milk in Tris-buffered saline containing 0.05% Tween-20. The membranes were then incubated with primary antibodies against Muc-2, Occ, Cla, ERK1/2 (CST-4695, Cell Signaling Technology, USA), and p-ERK1/2 (CST-4370, Cell Signaling Technology, USA). The cells were incubated overnight at 4 °C and then incubated with the secondary antibody for 2 h at 37 °C. Protein bands were detected using enhanced chemiluminescence and imaged using the Clinx Chemiluminescence Imaging System. The intensity of the β-actin protein band was used as an internal reference.
Data analysis
GraphPad Prism 9 (GraphPad Prism Software, San Diego, CA, USA) was used to analyze the data and construct the graphs. The data were confirmed to be normally distributed, and the homogeneity of variance was determined before performing independent sample t-tests, including calculating the mean and standard deviation of the data. The significance level was set at P < 0.05.




Comments (0)