Human subjects
This study was approved by the Medical Ethics Committee of Sun Yat-sen Memorial Hospital of Sun Yat-sen University (SYSKY-2023-342-01), and signed informed consent forms were obtained from each subject. We collected 60 patients who underwent surgery at Sun Yat-sen Memorial Hospital of Sun Yat-sen University due to cervical spondylosis and divided them into two groups: the Control group (n = 40) and the Hyperglycemia group (n = 20). We analyzed their preoperative CT scanning data and calculated the rate of HO that occurred in the ligamentum nuchae (OLN), posterior longitudinal ligament (OPLL), or ligamentum flavum (OLF). The calcified tendon was obtained from 12 patients with HO during surgery. We also collected the normal Achilles tendon from patients who received surgery (n = 5) and from DM patients who received surgery due to the diabetic foot (n = 5). The serums of the control patients (n = 10) and DM patients (n = 10) were collected for the ELISA test.
Mice
All mice (C57BL/6J) were purchased and bred at the Laboratory Animal Center of Sun Yat-sen University. The animal experiments were approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University (SYSU-IACUC-2023-000847). We used mice aged from 6 to 8 weeks for the DM model by feeding them with 60% high-fat fodder (HF60 fodder, Dyets) for two months and then exerted Achilles tenotomy to induce the occurrence of HO. We measured the blood glucose level using a glucometer, and a blood glucose higher than 33.3 mmol/L was displayed as High, and lower than 1.1 mmol/L as Low. Before the glucose tolerance test (GTT) and insulin tolerance test (ITT), the mice fasted for eight hours. For the GTT, we treated the mice with 2 g/kg glucose via intraperitoneal injection and the insulin level was measured using ELISA (E-EL-M2614c-96T, Elabscience). For ITT, we treated the mice with 1 U/kg insulin via intraperitoneal injection.
Histological analysis
The tissues were disserted and fixed in 4% paraformaldehyde for 48 h at 4 °C. After fixation, the tissues were embedded in paraffin and processed for paraffin sectioning at 5 μm thickness. H&E staining, Safranine O staining, and Masson trichrome staining were performed according to standard procedures. The slides were photographed under a light microscope (Olympus).
Immunohistochemistry
The slides were immunoreacted with anti-TMND (Abcam, ab203676, 1:250), anti-RUNX2 (Abcam, ab192256, 1:500), anti-OPN (Abcam, ab214050, 1:500), anti-p16 (Santa Cruz, sc-1661, 1:100), and anti-CXCL13 (Abcam, ab272874, 1:100) or anti-CXCR5 (Abcam, ab203212, 1:500). Further processing occurred according to standard procedures. The IHC score was calculated independently by experimenters blinded to the sample identity. Staining intensity was scored as follows: 0 (negative), 1 (weakly positive), 2 (moderately positive), and 3 (strongly positive). The percentage of positivity was also scored according to five categories: 0 (< 5%), 1 (5–25%), 2 (25–50%), 3 (50–75%), and 4 (> 75%). The value of the percentage positive score was multiplied by the staining intensity score to generate the final expression scores, which ranged from 0 to 12.
Isolation and culture of TSCs
The patellar tendon was isolated and washed using PBS, then cut into small pieces and digested with type I collagenase (3 mg/ml, Sigma-Aldrich) for 6 h at 37℃. The cells were then resuspended and seeded in a 6-well plate and cultured at 37 °C with 5% CO2. Low glucose DMEM (Gibco) supplemented with 1% penicillin–streptomycin solution (Gibco) and 10% fetal bovine serum (Gibco) were used as the culture medium. For osteogenic induction, the OIM was the culture medium supplemented with 20 mM β-glycerolphosphate, 50 mM ascorbic acid, and 1 nM dexamethasone for 7 or 14 days. The alkaline phosphatase (ALP) staining and alizarin red staining were performed according to standard procedures.
Cell Counting Kit-8 assay (CCK-8)
The TSCs were seeded at a density of 1 × 104 per well in 96-well plates and cultured in culture medium or high glucose DMEM supplemented with different concentrations of palmitic acid for 0, 12, 24, and 48 h. CCK-8 solution (Glpbio) was added to each well and incubated for 3 h, followed by measurement of the absorbance at 450 nm using a microplate reader (TECAN sunrise).
Immunoblotting analysis
We performed western blotting according to standard procedures. The following primary antibodies were used: anti-OCT4 (CST, #2750, 1:500), anti-NANOG (Abcam, ab109250, 1:500), anti-p21 (Abcam, ab109520, 1:1,000), anti-TNMD (Abcam, ab203676, 1:250), anti-RUNX2 (Abcam, ab192256, 1:1,000), anti-OPN (Abcam, ab214050, 1:500), anti-Cyclin D1(Santa Cruz, sc-8396, 1:500), anti-Cyclin B1 (Santa Cruz, sc-245, 1:500), anti-CXCL13 (Abcam, ab272874, 1:250), anti-CXCR5 (Abcam, ab203212, 1:500), anti-GAPDH (Proteintech, 60004-1, 1:2,000), or anti-β-ACTIN (Affinity, AF7018, 1:2,000). The secondary antibodies used were goat anti-rabbit IgG H&L (Abcam ab205718, 1:2,000) and goat anti-mouse IgG H&L (Abcam ab205719, 1:2,000). The original blot results are shown in Supplementary Figure S6.
Senescence-associated β-galactosidase (SA-β-gal) staining
SA-β-gal staining (Beyotime) was performed according to the manufacturer’s protocol. Briefly, the cells were washed with PBS and fixed in the SA-β-gal fixative for 15 min at room temperature, followed by incubation with SA-β-gal working solution at 37℃ overnight. After staining, three random fields of each group were imaged under an optical microscope, and the ratio of β-Gal + cells was calculated.
qPCR
Total RNA was extracted from cultured cells using TRIZOL reagent (Invitrogen), and cDNA was synthesized using 1,000 ng of total RNA with a Prime-Script RT reagent kit (TaKaRa) according to the manufacturer’s protocol. qPCR was performed to amplify the cDNA on a Light Cycler 480 Real-Time PCR system (Roche Light Cycler 480) using TB Green Premix Taq II (TaKaRa) and corresponding primers. The sequences of primers were acquired from PrimerBank [54], and the expected size of the amplicon in bp, the annealing temperatures, GenBank Accession numbers are listed in Supplementary Table 2. The 2−ΔΔCt method was used to calculate the relative expression levels, and the expression of Actb served as the internal control for normalization.
RNA-seq and analysis
Total RNA was extracted from TSCs cultured in low glucose or high fat, high glucose (HFHG), and libraries were generated using the VAHTS Stranded mRNA-seq Library Prep Kit for Illumina (New England Biolabs). Library quality was checked with a Bioptic Qsep100 Analyzer (Bioptic Inc.), and libraries were sequenced using NovaSeq 6000. Genes with a fold change > 2 and p-value < 0.05 were identified as differentially expressed genes and analyzed using DAVID Bioinformatics Resources. Four biological replicates in each group were included in the RNA-seq experiment in this study.
Bone CT scanning
The hindlimbs of the mice were dissected and fixed in 4% paraformaldehyde for 48 h at 4 °C, followed by storage in 70% ethanol for micro-CT scanning (SCANCO Medical AG). Quantitative volumetric measurements of HO were conducted on the tibia region.
CRISPR/sgRNA plasmid construction
The lenti-CRISPR v2 plasmid was available from Addgene (52,961). The targeted sgRNAs were inserted into the plasmid using BsmBI sites. The sgRNA sequences for Cxcl13 were as follows: sgRNA-1 (TCGGTCTAAACATCATAGAT, PAM: CGG) and sgRNA-2 (GGATTCAAGTTACGCCCCCT, PAM: GGG).
Vector transduction and lentivirus/adenovirus infection
Lentivirus infection was performed according to the manufacturer’s protocol of the GeneCopoeia Lenti-Pac kit. Briefly, HEK293T cells were plated in high-glucose DMEM (Gibco) with 10% FBS and 1% penicillin–streptomycin. The cells were co-transfected with 2.5 µg lenti-CRISPR plasmid and packing plasmids (pLP1, pLP2, and pLP-vsvg) using Lipofectamine 3000 (Thermo Fisher Scientific). After 8–14 h, the transfection medium was changed, and the medium with lentivirus was harvested after 48 h. The lentiviral supernatant was filtered through 0.45 μm filters to eliminate residual cells and cell fragments.
After the cells were attached to the dish with 70–80% confluency, the adenovirus or lentivirus was added to the non-FBS medium (109 PFU/ml). The same volume medium with 10% FBS was added to the cells, and the medium was changed after 24 h. The cells were cultured for another 24 h before the next step of the experiments.
Statistical analysis
Quantification was performed in at least three independent experimental groups. The analysis of all statistics was carried out using SPSS v13. We tested the values with the Kolmogorov–Smirnov test (K–S test) to determine the normally distributed, and the two-tailed Student’s t-test (normally distributed) or Mann–Whitney U test (not normally distributed) was used between two groups to determine the significance. A one-way ANOVA (normally distributed) with Tukey’s post-hoc test was used to compare differences between multiple groups. In all cases, a p-value of less than 0.05 was considered significant, and the results were presented as mean ± standard deviation (SD).

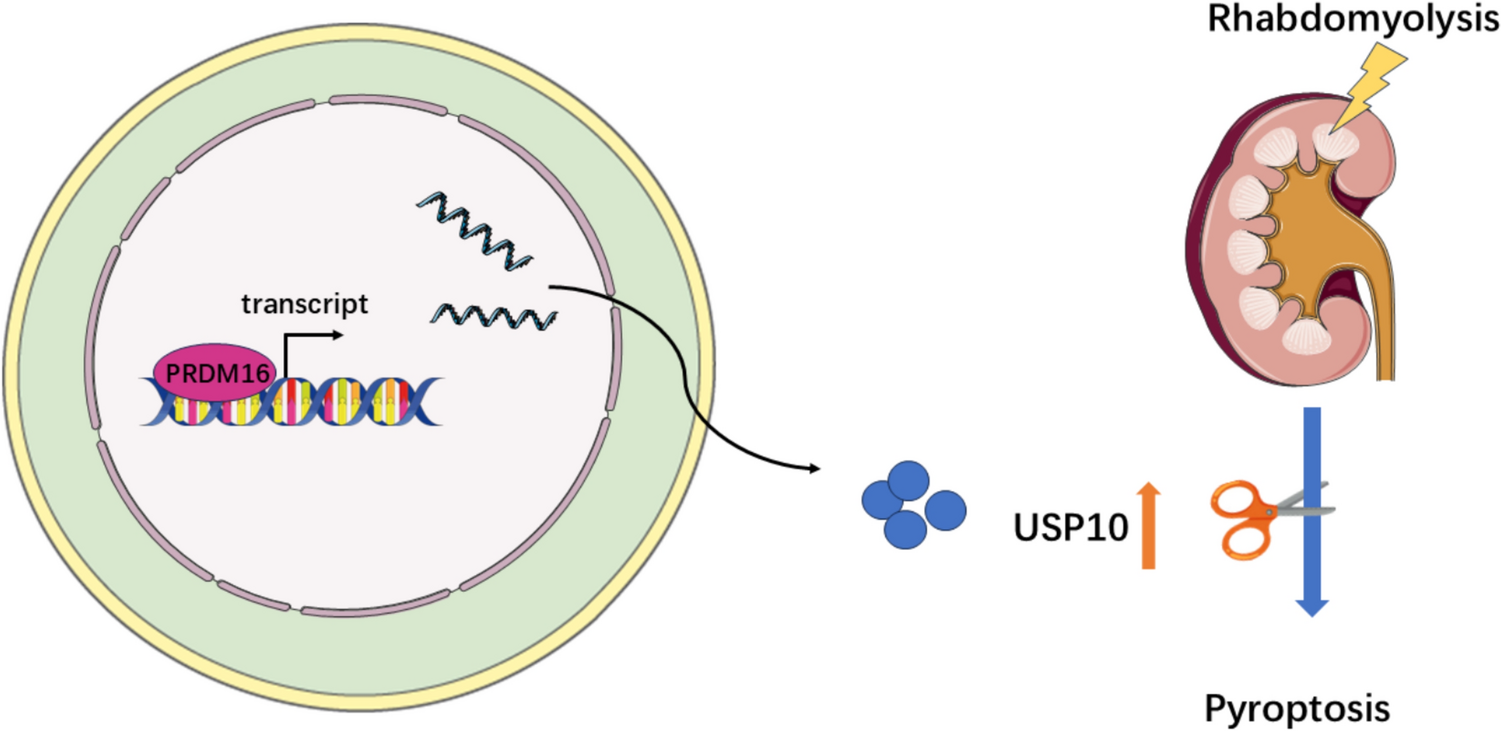

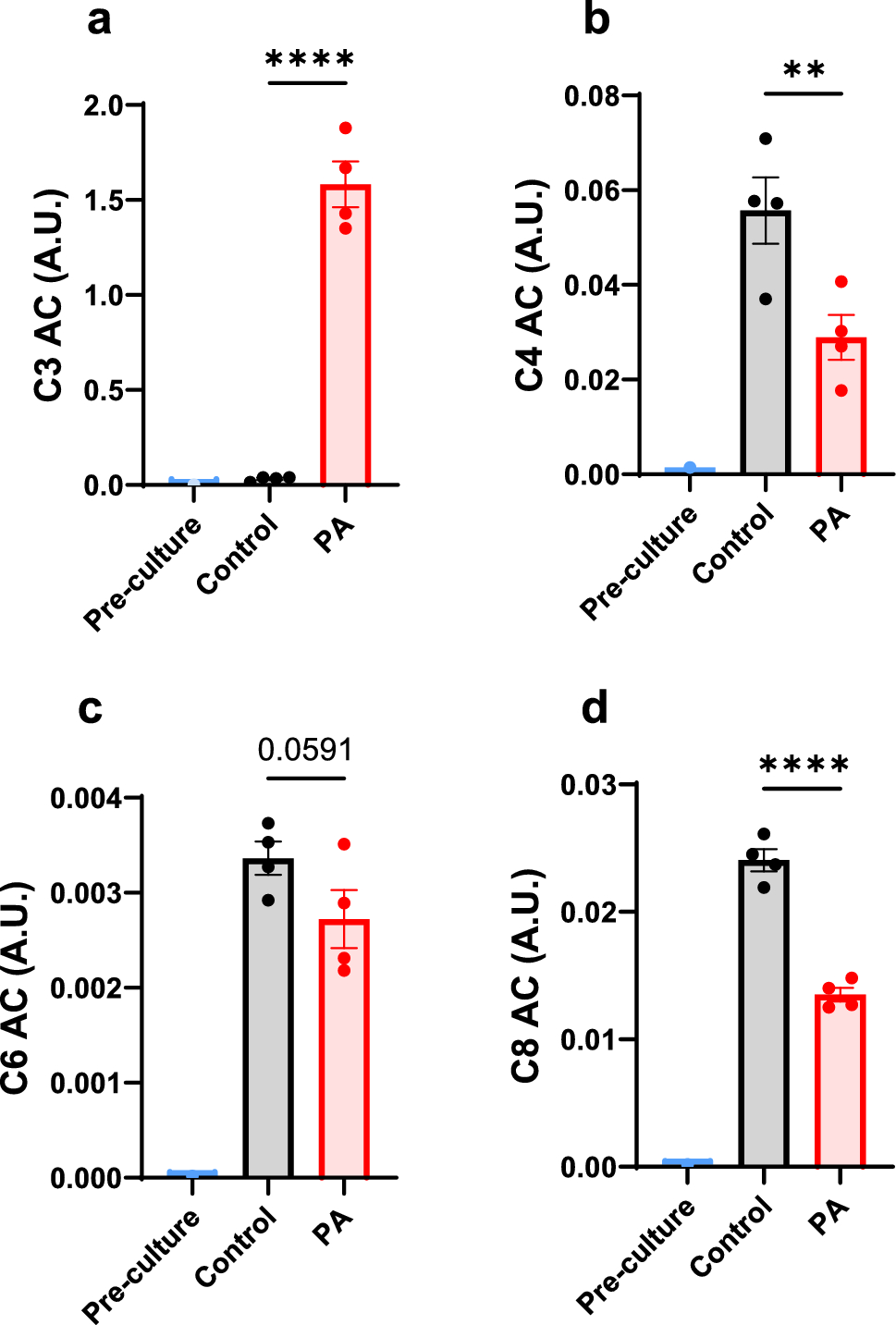
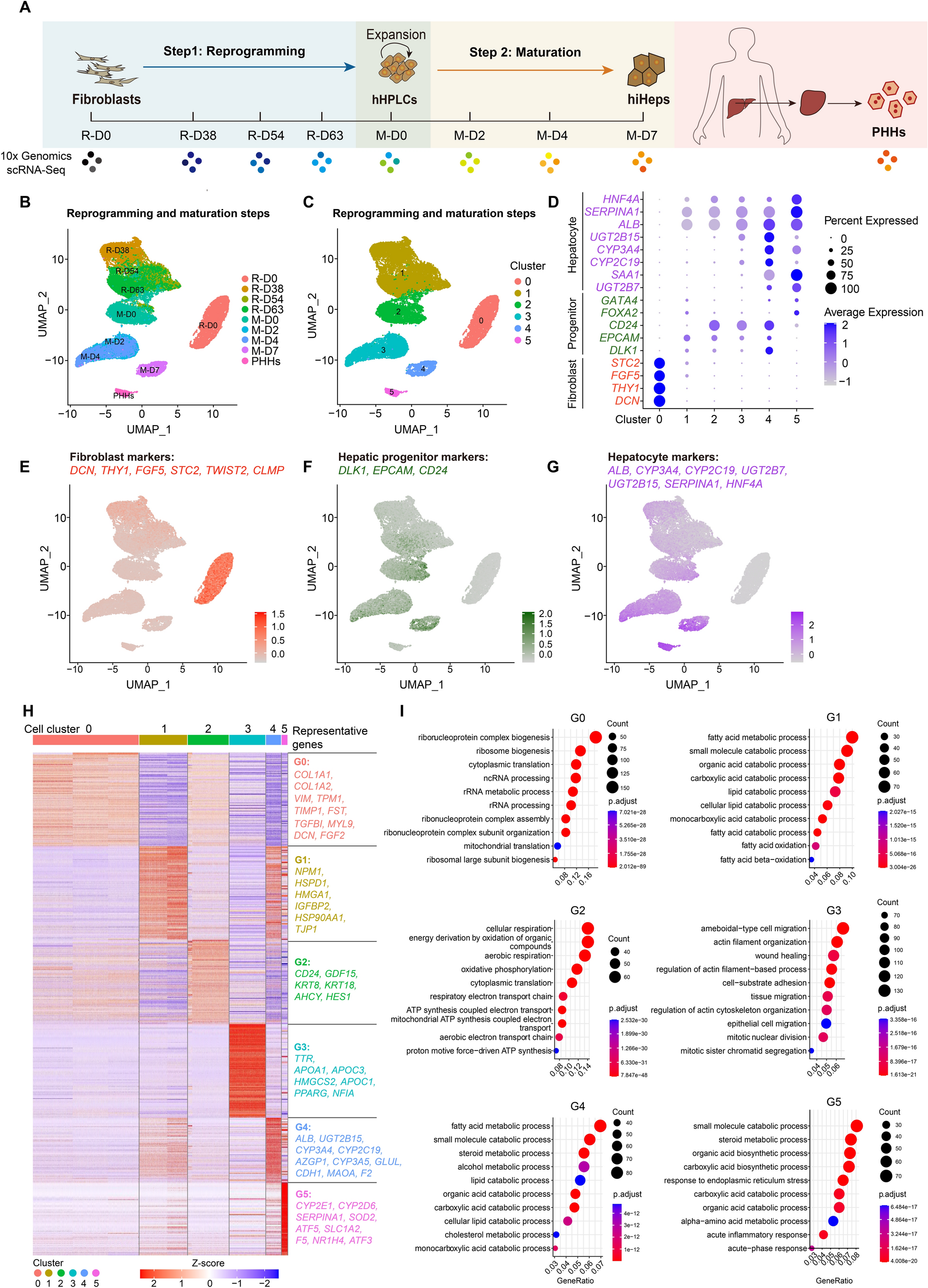
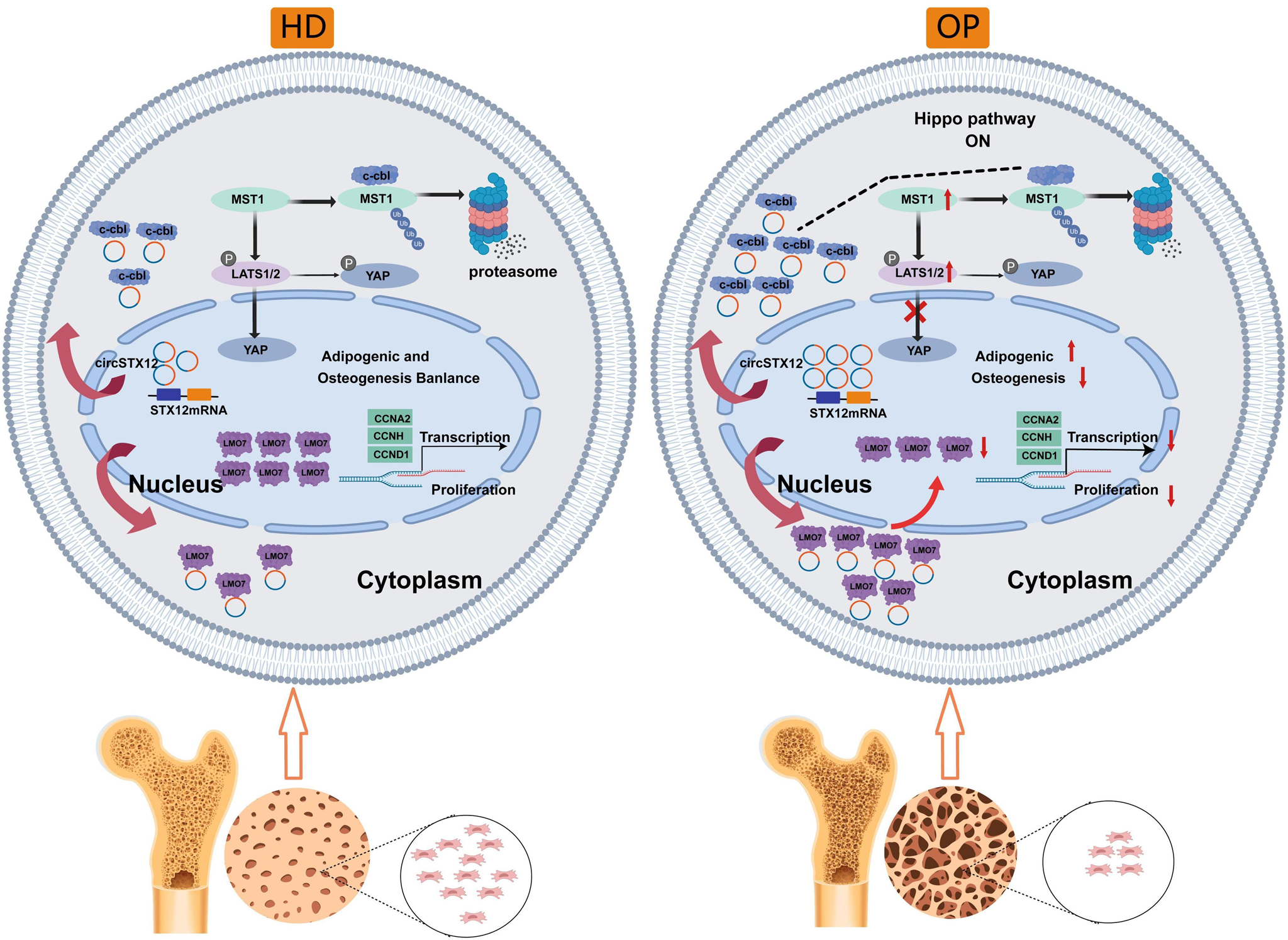

Comments (0)