
Remember me
TP53 mutations (TP53Mut) define the most rapidly fatal AML subtype [1, 2] (Supplementary Fig. S1A). We used AML datasets (Beat AML and TCGA LAML [3,4,5,6] (Supplementary Tables S1–3), to define the gene expression profile (GEP) of TP53Mut AML. The diagnostic, relapsed, and refractory TP53Mut cases in Beat AML were transcriptionally similar to those in the TCGA (which includes only diagnostic cases, Fig. 1A). Neither principal component analysis (PCA) nor hierarchical clustering detected significant clustering according to TP53 status (Supplementary Fig. S1B–D). Therefore, we used logistic regression with ridge regularization to learn the GEP features that define TP53Mut AML. We separated the Beat AML dataset into training (60% of the cases) and test datasets (40% of the cases) and trained our model to classify TP53Mut cases. The trained classifier model was highly accurate in detecting TP53Mut cases in the test dataset (Supplementary Fig. S1E). As validation, we found that the model was also highly accurate in classifying TP53Mut cases in the TCGA (Supplementary Fig. S1E).
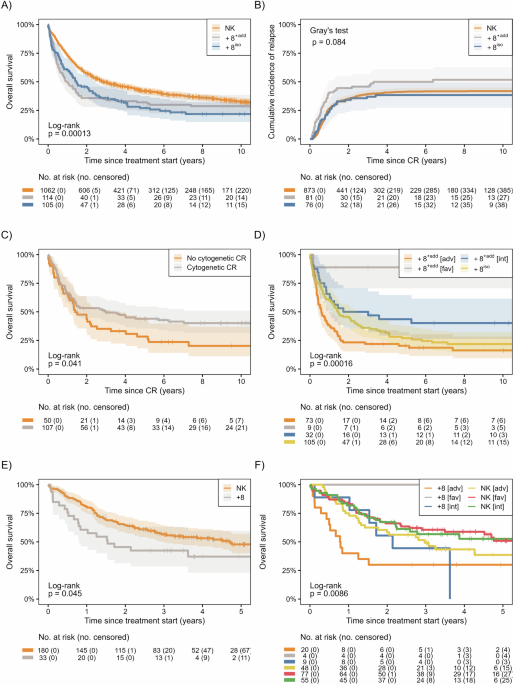
Fig. 1: TP53Mut-like AML: a subset of TP53WT AMLs that share GEP features and poor clinical outcomes with TP53Mut AML.
A Principal Component Analysis (PCA) of TP53Mut samples in the Beat AML and TCGA LAML dataset (Beat AML: TP53Mut n = 36; 19 diagnostic, 2 relapse and 15 residual cases, TCGA LAML: TP53Mut n = 15; all diagnostic cases). B–E We used a ridge regression model as a classifier to classify TP53Mut AML and TP53Mut ridge score reflects how closely a GEP resembles that of TP53Mut AML GEPs. As expected, TP53Mut AMLs have high TP53Mut ridge scores and poor OS in both Beat AML and TCGA LAML datasets (Supplementary Fig. 2A). B TP53Mut ridge scores are plotted versus overall survival in the diagnostic samples in Beat AML dataset. C Kaplan–Meier survival curves of diagnostic samples in the Beat AML dataset. D TP53Mut-like ridge scores are plotted versus survival in the TCGA LAML validation dataset. E Kaplan–Meier survival curves of samples in the TCGA LAML dataset. C, E P values reflect pairwise comparisons between TP53Mut, TP53Mut-like and TP53WT samples. Log-rank test was used to calculate P values. Median survival: Beat AML TP53Mut: 167 days (0.46 years), Beat AML TP53Mut-like: 204 days (0.56 years), Beat AML TP53WT: 861 days (2.36 years); TCGA LAML TP53Mut: 130 days (0.36 years), TCGA LAML TP53Mut-like: 335 days (0.92 years), TCGA LAML TP53WT: 800 days (2.19 years). Beat AML: TP53Mut n = 36 (19 diagnostic samples), TP53Mut-like n = 40 (26 diagnostic samples) and TP53WT n = 335 (223 diagnostic samples). TCGA LAML (all diagnostic samples): TP53Mut n = 15, TP53Mut-like n = 23 and TP53WT n = 140. F The fraction of TP53Mut and TP53Mut-like AMLs in Beat AML and TCGA LAML datasets. G PCA of samples in the Beat AML and TCGA LAML dataset (Beat AML: TP53Mut: biallelic: n = 29, monoallelic: n = 7, TP53Mut-like n = 40, TP53WT n = 327; TCGA LAML: TP53Mut: biallelic: n = 15, monoallelic: 0, TP53Mut-like n = 23, TP53WT n = 140). Fraction of all samples in each TP53 category that harbor H 17p alterations by karyotype or I TP53 locus alterations by copy number array, including amplifications and deletions (copy number array data is not available in the Beat AML). J Bone marrow blast percentage, and K white blood cell counts were plotted for each TP53Mut, TP53Mut-like, and TP53WT AML diagnostic sample in the Beat AML dataset. Horizontal red bars indicate the mean values. Error bars represent standard error of the mean. Unpaired Student t-test was used to calculate P values for each comparison. Benjamini-Hochberg method was used to correct for multiple hypothesis testing and to calculate the false discovery rate (FDR). Detailed statistical data (FDR values for each comparison) are listed in Supplementary Table S11. L Fraction of diagnostic cases that are TP53Mut-like in each ELN 2022 risk category. Favorable risk: Beat AML: n = 3 (3.6%); TCGA LAML: n = 3 (4.8%). Intermediate risk: Beat AML: n = 4 (6.0%); TCGA LAML: n = 6 (13.0%). Adverse risk: Beat AML: n = 18 (19.8%); TCGA LAML: n = 13 cases (28.9%).
Strikingly, we noticed a subset of TP53WT cases with high ridge scores (indicating high similarity to the TP53Mut GEP) in the Beat AML. High-scoring TP53WT cases had low overall survival (OS, Supplementary Fig. S2A). We defined the TP53WT samples in the top 10% of ridge scores (n = 40) as TP53Mut-like since these cases transcriptionally and prognostically resemble TP53Mut cases (Fig. 1B, C). To detect whether the TCGA also harbors TP53Mut-like cases, we trained a new, complementary ridge regression model using the TP53WT cases in the Beat AML (excluding TP53Mut cases). This new model was highly sensitive and specific in classifying the held-out patients in the Beat AML (Supplementary Fig. S2B, D). We applied this TP53Mut-like model to the TCGA and validated that high TP53Mut-like ridge scores identify a subset of TP53WT patients with poor OS in TCGA as well (Fig. 1D–F, Supplementary Fig. S2EF).
Beat AML and TCGA AMLs vary by subtype and disease stage. TCGA includes only diagnostic, de novo AMLs [3] while Beat AML includes all subtypes at any disease stage disease [5, 6] (Fig. 1F, Supplementary Tables S1 and 2). Therefore, we confirmed that disease stage does not impact the transcriptional landscape of TP53Mut and TP53Mut-like cases across both datasets (Supplementary Fig. S3A–C). To further validate our findings, we reversed our analysis and trained a new ridge regression model on TCGA cases and tested this model on the Beat AML dataset. The TCGA-derived model shows high accuracy in detecting TP53Mut cases in Beat AML regardless of disease stage (Supplementary Fig. S3D–F, Supplementary Table S4). These data confirm that the TP53Mut GEP is consistent across disease stages.
Next, we assessed the impact of other potential confounding features (Supplementary Fig. S4). We compared ridge scores of de novo, secondary and treatment-related TP53Mut and TP53Mut-like AMLs and found no significant differences, suggesting that ridge scores are not a reflection of these AML subtypes.
We also compared how TP53 locus status impacts ridge scores. Monoallelic and biallelic TP53 altered AMLs were not distinguishable based on PCA (Fig. 1G, Supplementary Table S5). TP53 allele status did not correlate with ridge score (Supplementary Fig. S5A–C). Furthermore, 15–17% of TP53Mut-like cases harbor 17p alterations but these alterations did not impact OS (Fig. 1H, I, Supplementary Fig. S5D–I, Supplementary Table S6–S10). Therefore, TP53 locus deletion is not sufficient to induce a TP53Mut-like phenotype.
We next investigated whether the TP53Mut and TP53Mut-like AMLs share similar clinical parameters. TP53Mut and TP53Mut-like patients have significantly lower bone marrow blasts, white blood cell counts, and are older than TP53WT AMLs in the Beat AML dataset. We found similar trends in the TCGA, but with variable statistical significance likely due to smaller sample sizes (Fig. 1J, K, Supplementary Fig. S6A–D, Supplementary Table S11). The ridge score was not correlated with leukemia burden (Supplementary Fig. S6E). Together, these data suggest that TP53Mut-like AML share the distinct clinical and biological characteristics of TP53Mut AML.
We found TP53Mut-like cases in all ELN risk categories [2] in both datasets (Fig. 1L, Supplementary Fig. S7A). As expected, the largest fraction of TP53Mut-like AMLs was adverse risk. However, TP53Mut-like cases represent 3.6–4.8% of the favorable risk cases and 6.0–13.0% of intermediate risk cases. TP53Mut-like cases have a trend towards inferior survival in both the favorable and adverse risk categories, but the number of cases was small (Supplementary Fig. S7B, Supplementary Table S12).
Next, we analyzed the ex vivo drug sensitivity profiles in the Beat AML. When compared to the TP53WT cases, the TP53Mut-like cases resemble TP53Mut cases, showing resistance to most drugs (Fig. 2A, Supplementary Fig. S8). Like TP53Mut AML, TP53Mut-like samples are highly resistant to venetoclax, a standard-of-care AML therapy (Fig. 2B). Interestingly, the resistance profile of TP53Mut-like samples did not fully recapitulate that of the TP53Mut samples but the differences between these samples were not statistically significant (Supplementary Fig. S8).
Fig. 2: TP53Mut-like AML and TP53Mut AML share drug-resistant patterns and gene expression profiles and can be identified using 25-gene signature.
A, B Ex vivo drug sensitivity data generated from 122 small molecule inhibitors in the Beat AML dataset. Area under the curve (AUC) values were Z-score transformed and multiplied by −1 to generate AUC Z-scores, raw, untransformed AUC data is displayed in Supplementary Fig. S8A. High Z-score indicates drug sensitivity. A Heatmap of entire dataset. B Venetoclax sensitivity of TP53Mut, TP53Mut-like and TP53WT samples. Unpaired Student t-test was used to compare the average differences in AUCs between TP53Mut or TP53Mut-like to TP53WT samples. Venetoclax was not routinely used to treat the patients in either database, which precludes analysis of patient treatment response. Multiple hypothesis testing was corrected using the Benjamini-Hochberg method to calculate FDR. C GSEA was performed to compare TP53Mut and TP53WT samples (red and blue), and TP53Mut-like and TP53WT samples (orange and skyblue). Gene sets displayed are those that are significantly enriched in both Beat AML and TCGA datasets, based on concordant normalized enrichment scores (NES) and FDR < 0.05 in both datasets independently. Genes encoding cell surface markers that are differentially expressed between D TP53Mut and TP53WT, and E TP53Mut-like and TP53WT samples. Genes encoding cell surface markers that are displayed are those concordantly differentially expressed in both the Beat AML and TCGA LAML datasets with an FDR < 0.05 in each dataset. Data is displayed as log2 transformed CPM expression values that were mean-centered to generate z-scores. Genes that are concordantly shared between TP53Mut and TP53Mut-like (D and E) are marked with red (up-regulated) and blue (down-regulated) asterisks. Beat AML data is shown (TCGA data is shown in Supplementary Fig. S10). F 25-gene signature that defines TP53Mut-like AML. Expression values of 25-gene signature genes in Beat AML samples were shown. CPM values were log2 transformed and Z-score converted. These 25 core genes are a subset of the full TP53Mut-like signature genes (listed in Supplementary Table S16). We performed 100 iterations of this analysis, genes that were recurrently identified across multiple iterations are listed in Supplementary Table S16. In addition, we queried whether our 25-gene signature was shared with known TP53 target genes or previously published TP53Mut AML gene signatures but our 25-gene signature did not overlap with them (Supplementary Table S17).
We performed gene set enrichment analysis [7] on the differences between TP53Mut or TP53Mut-like samples to TP53WT samples (each comparison was performed separately in each dataset). We identified overlapping gene sets as those with significant and concordant enrichment in both datasets (Fig. 2C, Supplementary Fig. 9A, Supplementary Tables S13 and S14). Notably, both TP53Mut and TP53Mut-like AMLs were strongly enriched with NFκB, inflammatory and stem cell pathways and EZH2 targets. In contrast, TP53Mut and TP53Mut-like AMLs displayed negative enrichment (downregulation) of oxidative phosphorylation and mitochondrial pathways. Using Ingenuity Pathway Analysis, we also found that TP53Mut-like and TP53Mut cases share activation of NFκB and inflammatory regulators (Supplementary Table S15), consistent with reports that chronic inflammation is associated with TP53Mut leukemic progression [8].
Next, we searched for genes that encode protein markers of TP53Mut and TP53Mut-like cases. In comparing TP53Mut and TP53WT cases, 13 genes that encode cell surface proteins that were significantly and concordantly differentially expressed in both datasets (Fig. 2D, Supplementary Fig. S10, Supplementary Table S13). In comparing TP53Mut-like to TP53WT cases, 16 cell surface marker-encoding genes were significantly and concordantly differentially expressed in both datasets (Fig. 2E, Supplementary Fig. S10, Supplementary Table S13). Among these genes, five genes were identified in both comparisons. If validated at the protein level, these cell surface markers offer potential therapy targets for these AML subsets.
Next, we asked whether a concise gene signature could be used to identify TP53Mut-like AML across datasets. We used elastic net regression, which results in sparser models [9] and is better suited to identify a concise gene signature. We performed multiple rounds of elastic net optimization and identified 25 core genes that accurately classify TP53Mut-like AML. A new ridge regression model, built with those 25 genes, showed high classification accuracy for TP53Mut-like AMLs in both datasets (Fig. 2F, Supplementary Fig. S11, Supplementary Table S16). This 25-gene signature can be used as a diagnostic assay to identify TP53Mut-like AMLs.
In summary, we used GEPs and a machine learning classifier to define TP53Mut-like AML, a novel subtype of TP53WT AML that transcriptionally and prognostically phenocopies TP53Mut AML. Notably, this subset is imperceptible using traditional unsupervised clustering methods and demonstrates the power of supervised machine learning approaches. TP53Mut-like AMLs share poor survival rates, distinct clinical parameters, and biological pathways with TP53Mut AML. TP53Mut-like AMLs also display wide-spread in vitro drug resistance. Finally, we discovered a 25-gene signature that can be used to identify TP53Mut-like AMLs.
Mutational and cytogenetic profiling are the most common molecular approaches to classify malignancies. However, the functional insights provided by transcriptional profiling can reveal clinically distinct subsets that are not detected using these methods. The GEP of acute lymphoblastic leukemia (ALL) identified a subset of ALL that resembles Philadelphia chromosome positive (Ph+) ALL. Like the TP53Mut-like AMLs we describe here, Ph-like ALLs share poor prognostic features with Ph+ ALL, including high relapse rates [10, 11] and represent a distinct clinical entity that requires more aggressive consolidation therapy [12]. GEP has also defined novel disease subtypes in lymphoma [13] and breast cancer [14].
TP53Mut and TP53Mut-like AMLs uniquely express cell surface marker genes. Future work to validate the cell surface protein profile of these AMLs could include these proteins. Notably, CD99, which is a candidate therapeutic target in AML [15], is downregulated in both TP53Mut and TP53Mut-like cases. Once validated, the protein products of these genes could provide targets for immunotherapy or serve as labels to quickly identify these cases clinically.
Rapid RT-PCR assays are routinely used in the diagnostic workflow for acute leukemia to test for PML-RARA, BCR-ABL, and mutant FLT3. Our 25-gene assay would fit within this standard workflow without significantly increasing the turnaround time: RT-PCR assays can be readily multiplexed and have a rapid turnaround time that can be resulted within hours. Future work could validate our 25-gene signature in a prospective cohort of patients.
Clinical trials are underway to evaluate promising novel approaches in TP53Mut AML. Our data suggests that TP53Mut-like patients might benefit from the same treatment strategies as TP53Mut AML. The overlap between deregulated pathways in TP53Mut and TP53Mut-like cases might suggest that both subsets might benefit from similar therapies. Future work could test whether targeting these pathways could offer clinical benefit in TP53Mut and TP53Mut-like AML. Our 25-gene signature could be used to identify such patients for clinical trial inclusion and expand the number of patients eligible for such clinical trials.
Comments (0)