Samples
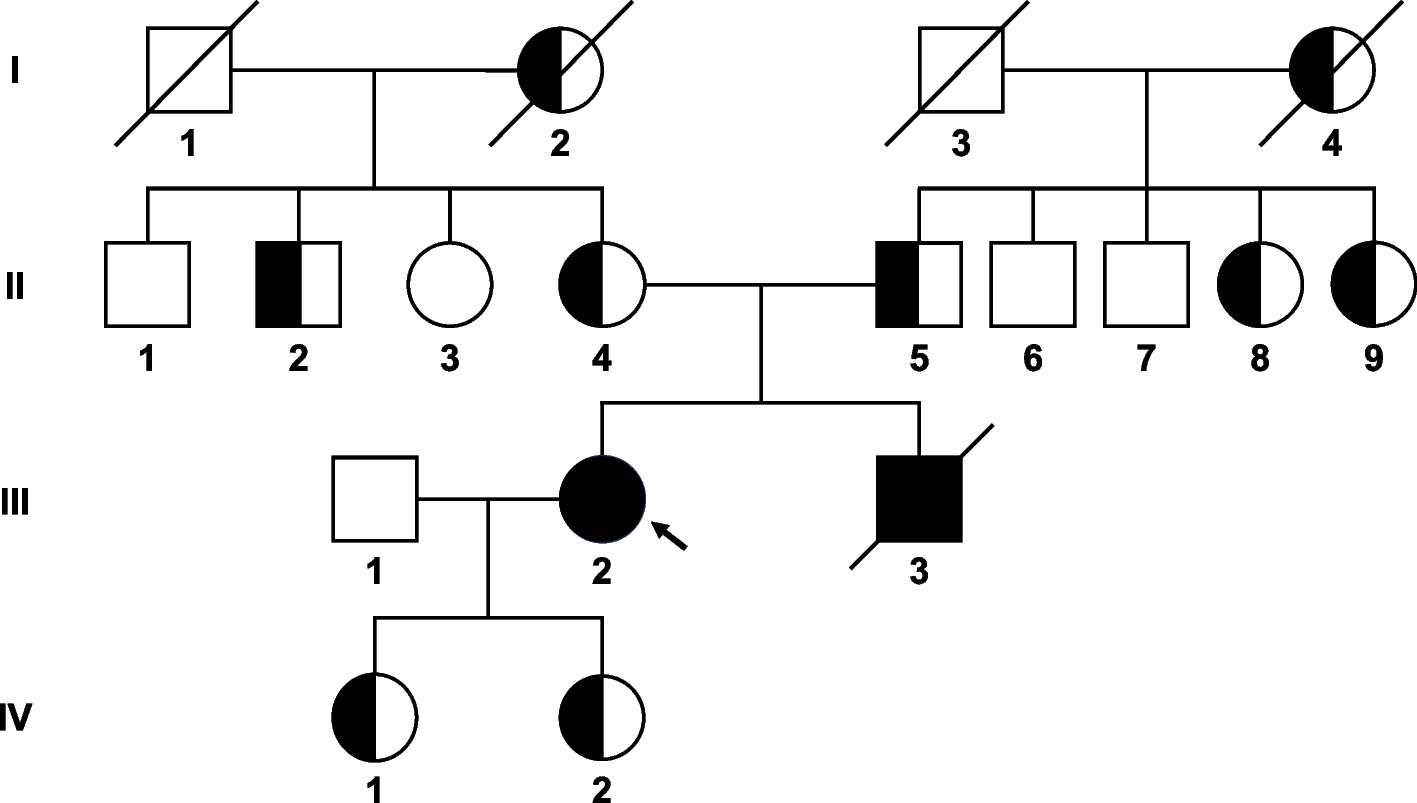
The here discussed patients were two sisters who were diagnosed with AT deficiency after multiple thromboembolic events. Patient 1, a 64-year-old woman, suffered from a bilateral deep vein thrombosis (DVT) as a teenager with recurrence after a temporary change in her anticoagulation treatment from phenprocoumon to acetylsalicylic acid. The treatment regimen was switched back to phenprocoumon after the DVT. Her sister (patient 2), a 65-year-old woman, had multiple DVT events in her right lower leg and pulmonary embolism as a teenager. The patient also developed a DVT in her right leg at the age of 27 during pregnancy eight weeks before delivery. The intake of an oral contraceptive during the time of their first thrombotic events was postulated as a possible triggering factor. In addition, both patients have been diagnosed with a heterozygous factor V Leiden (FVL) mutation. According to the family history, the patients´ father was diagnosed with AT deficiency. No statement could be made from the patients as to whether their parents suffered from thromboembolic events. Additionally, the patients could not provide any information regarding the heredity of the FVL mutation within their family.
Blood samples from healthy blood donors of the blood donation center of the Clinical Department of Transfusion Medicine and Hemostaseology in Erlangen served as control material within the study. The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the Friedrich-Alexander-University Erlangen-Nürnberg (FAU) (#477_20 B, #357_19 B, #343_18 B). Written informed consent was obtained from all participants of the study.
Determination of AT parameters
AT activity and AT antigen levels were analyzed with a STA Max instrument (Diagnostica Stago S.A.S., Asnières-sur-Seine, France), using the STACHROM AT III Kit for activity and the LIATEST AT III kit for antigen levels (Diagnostica Stago S.A.S., Asnières-sur-Seine, France).
DNA isolation
Genomic DNA was automatically isolated from whole blood and quantified using commercial kits (NucleoSpin® 8 Blood Core Kit, Macherey Nagel, Düren, Germany for isolation, QuantiFluor® dsDNA System, Promega, Madison, WI, USA for quantification) and a Hamilton Microlab Starlet robot (Reno, NV, USA) and microplate fluorometer (Berthold, Bad Wildbad, Germany).
Next generation sequencing
The CDS (according to transcript NM_000488.3) and adjacent intron regions of the SERPINC1 gene were sequenced with an Illumina amplicon-based gene panel (AmpliSeq™ Custom DNA Panel for Illumina®) on a MiSeq machine (Illumina®, San Diego, CA, USA) in accordance to the manufacturer’s instructions. The custom DNA panel enabled CDS and adjacent intron regions sequencing of the following genes: F7, F9, F13A1, F13B, F11, FGA, FGB, FGG, PROC, PROS1, SERPINC1 and VWF [6].
Next generation sequencing data was thereafter analyzed with the SeqNext Software version 5.2.0 (JSI Medical Systems, Ettenheim, Germany).
Database submission
The c.1274dupC frameshift mutation was submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar; accession SCV002520629).
Quantitative mutation screening
To gage the prevalence of the c.1247dupC frameshift mutation within the general population, 358 healthy individuals were screened with a sequence-specific primer polymerase chain reaction (SSP-PCR) as described before [6]. Oligonucleotides were obtained from Merck (Darmstadt, Germany). Primer combinations consisted of the following: SERPINC1g+13006f (5’-AGTACCTTACATTCTCTGCATGA-3’) and SERPINC1g+13243r-WT (5’-CAATCACAACAGCGGTACTTGC-3’) for the SERPINC1 WT allele (resulting in a 238 bp fragment) and SERPINC1g+13006f and SERPINC1g+13243r-SNP (5’-CAATCACAACAGCGGTACTTGG-3’) for detecting the SERPINC1 c.1247dupC frameshift mutation (resulting in a 239 bp fragment). The co-amplification of the human growth hormone gene (GH1) with primers GH1g+334f (5’-TGCCTTCCCAACCATTCCCTTA-3’) and GH1g+768r (5’-CCACTCACGGATTTCTGTTGTGTTTC-3’), resulting in a 434 bp fragment, served as an internal control.
Construct generation
Plasmids for HEK293T cell transfection were created using Gibson Assembly [7], according to the manufacturer’s instruction manual (Gibson Assembly® Master Mix/Gibson Assembly® Cloning Kit NEB, New England Biolabs, Ipswich, MA, USA). Oligonucleotides for Gibson Assembly were designed using NEBuilder Assembly Tool (https://nebuilder.neb.com/#!/) and were manufactured by Merck (Darmstadt, Germany). Vector pcDNA3.1( +)/Puro/FA/GFP-LL5BIP [kindly provided from Tomasz Prószyński (Addgene plasmid # 112829; http://n2t.net/addgene:112829; RRID:Addgene_112829)] was used for the expression of the GFP fusion. For plasmid pMR01 (SERPINC1-GFP) insert sequences and vector fragments with complementary overhangs were amplified by PCR using primers (and templates) as follows:
SERPINC1 CDS fragment: MR01-SERPINC1_fwd (5’-GTACCGAGCTCGGATCCATGTATTCCAATGTGATAGGAACTGTAACCTCTG-3’), MR01-SERPINC1_rev (5’-CACCATACCGCTACCGCCGCCGCTGCCACCCTTAACACAAGGGTTGGCTACTCTGC-3’) (human antithrombin III cDNA ORF Clone, obtained from SinoBiological, Beijing, China)
GFP CDS fragment: MR01-GFP_fwd (5’-GTTAAGGGTGGCAGCGGCGGCGGTAGCGGTATGGTGAGCAAGGGCGAGGAG-3’), MR01-GFP_rev (5’-GCACAGTCGAGGCTGATCACTTGTACAGCTCGTCCATGCCG-3’) (pcDNA3.1(+)/Puro/FA/GFP-LL5BIP)
vector pcDNA3.1 backbone: pcDNA3.1_fwd (5’-GGACGAGCTGTACAAGTGATCAGCCTCGACTGTGCCTTCTAG-3’), pcDNA3.1_rev (5’-CTATCACATTGGAATACATGGATCCGAGCTCGGTACCAAG-3’ (pcDNA3.1(+)/Puro/FA/GFP-LL5BIP).
For plasmid pMR02 (SERPINC1c.1247dupC-GFP) the c.1247dupC mutation was introduced by site-directed mutagenesis using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) [8], with the primer combination M1-pBWCS2_DupC_fwd (5’-GCAGCTGCCAAGTACCGCTGTTGTG-3’) and M1-pBWCS2_DupC_rev (5’-CGGTACTTGGCAGCTGCTTCACTG-3’). To deal with the resulting frameshift that also affects the CDS of GFP, SERPINC1 c.1247dupC CDS was amplified with oligonucleotides MR01-SERPINC1_fwd (5’-GTACCGAGCTCGGATCCATGTATTCCAATGTGATAGGAACTGTAACCTCTG-3’) and MR02-ohne Stop_rev (5’-GCTACCGCCGCCGCTGCCACCACACAAGGGTTGGCTACTCTGCC-3’) followed by the assembly into the pMR01 backbone (obtained with primer combination MR02-GGSG-EGFP_fwd: 5’-GGTGGCAGCGGCGG-3’ and MR01-pcDNA3.1_rev: 5’-CTATCACATTGGAATACATGGATCCGAGCTCGGTACCAAG-3’). During the PCR amplification for the Gibson Assembly a 2 × GGSG linker was introduced in front of the GFP in both pMR01 and pMR02.
The HA tag fusion vector was generated by digesting pcDNA3.1( +)/Puro/FA/GFP-LL5BIP with HindIII and XbaI to remove the GFP-LL5BIP cassette. Oligonucleotides encoding a 2 × GGSG linker and a 3 × HA tag (HindIII-GGSG-3xHA-Tag-XbaI_fwd: 5’-CTAGATCAAGCATAGTCAGGTACGTCATAAGGGTAAGATCCAGCATAGTCAGGTACGTCATAAGGGTAAGATCCAGCATAGTCAGGTACGTCATAAGGGTAACCGCTACCGCCA-3’ and HindIII-GGSG-3xHA-Tag-XbaI_rev: 5’-CTAGATCAAGCATAGTCAGGTACGTCATAAGGGTAAGATCCAGCATAGTCAGGTACGTCATAAGGGTAAGATCCAGCATAGTCAGGTACGTCATAAGGGTAACCGCTACCGCCA-3’) were annealed, leading to a double-stranded DNA fragment with HindIII and XbaI overhangs. This fragment was ligated into the HindIII/XbaI digested pcDNA3.1 backbone, resulting in the HA tag expression vector pBWCS3.
Insert and vector fragments for plasmid pMR03 (SERPINC1-HA) were amplified using primers (and template) as follows:
SERPINC1 CDS fragment: MR03_SERPINC-3xHA_fwd (5’- GCTAGCGTTTAAACTTAAGCTTATGTATTCCAATGTGATAGGAACTGTAACCTCTG-3’), pMR03_rev (5’-CATAAGGGTAACCGCTACCGCCGCCGCTGCCACCCTTAACACAAGGGTTGGCTACTCTGC-3’) (pMR01)
vector pBWCS3 backbone: pBWCS3-GGSG-3xHA_fwd (5’-GGCGGTAGCGGTTACCCTTATG-3’), pBWCS3-GGSG-3xHA_rev (5’-AAGCTTAAGTTTAAACGCTAGCCAGCTTG-3’) (pBWCS3).
For plasmid pMR04 (SERPINC1c.1247dupC-HA) the insert and vector fragments were amplified by PCR using the following primers (and template):
SERPINC1 c.1247dupC: MR03_SERPINC-3xHA_fwd (5’-GCTAGCGTTTAAACTTAAGCTTATGTATTCCAATGTGATAGGAACTGTAACCTCTG-3’), pMR04_rev (5’-CATAAGGGTAACCGCTACCGCCGCCGCTGCC ACCACACAAGGGTTGGCTACTCTGC-3’) (pMR02)
vector pBWCS3 backbone: pBWCS3-GGSG-3xHA_fwd (5’-GGCGGTAGCGGTTACCCTTATG-3’), pBWCS3-GGSG-3xHA_rev (5’-AAGCTTAAGTTTAAACGCTAGCCAGCTTG-3’) (pBWCS3).
Plasmid sequences were validated and confirmed by Sanger sequencing (Eurofins Genomics Germany GmbH, Ebersberg, Germany).
HEK293T clone generation
HEK293T cells (ACC 635, DSMZ, Braunschweig, Germany) were grown in DMEM (#D6429, Sigma-Aldrich, Taufkirchen, Germany) supplemented with 10% FCS (anprotec, Bruckberg, Germany), 1% GlutaMAX (Thermo Fisher Scientific, Waltham, MA, USA), 1% Penicillin–Streptomycin (Sigma-Aldrich, Taufkirchen, Germany) at 37°C and 5% CO2 in a humidified incubator. Cells were transfected with TransIT®-293 Reagent (Mirus Bio LLC, Madison, WI, USA) following the manufacturer´s recommendations. After 24 h the medium was replaced with fresh medium containing 5 µg/ml Puromycin (Carl Roth, Karlsruhe) to select the transfected cells. Clones were picked after 1–2 weeks and analyzed for the presence of the respective tag by flow cytometry (CytoFLEX S, Beckman Coulter, Brea, CA, USA). HEK293T cells transfected with pMR01 and pMR02 were analyzed for direct GFP expression while HA-tag expression of HEK293T cells transfected with pMR03 and pMR04 was assessed by intracellular staining in accordance to the manufacturer’s protocol (HA Tag Monoclonal antibody Dy550, #26183D550, Life Technologies, Carlsbad, CA, USA; eBioscience™ FOXP3-staining kit, #00–5523-00, Thermo Fisher Scientific, Waltham, MA, USA). Up to six cell lines were picked for each plasmid transfection and used for subsequent confocal laser scanning microscopy and Western blot.
Prior to microscopic analysis, HEK293T cells expressing plasmid pMR01 or pMR02 were transfected prior to imaging with the ER-Tracker™ Red (glibenclamide BODIPY® TR) (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min following the manufacturer´s recommendations.
Confocal laser scanning microscopy
A Leica TCS SP8 confocal laser scanning microscope (Leica, Wetzlar, Germany) was used for imaging with 488 nm (GFP) and 552 nm (RFP) laser light for excitation. GFP fluorescence was detected in a window ranging from 495–520 nm and RFP fluorescence in a window ranging from 570–595 nm. Data processing was performed with the Leica Application Suite X software version 2.0.1.14392 (Leica, Wetzlar, Germany).
Western blotting
In order to quantify protein content and secretion, respective HEK293T clones were cultured at 2 × 106–1 × 107 cells/ml in serum-free culture medium (#14571C, EX-CELL®, Merck, Darmstadt, Germany) without antibiotics for 48 h. Cells were subsequently harvested by resuspension and centrifuged at 300 g for 5 min. Supernatants were collected, mixed with the cOmplete™ Mini protease inhibitor-cocktail (Merck, Darmstadt, Germany) and subsequently stored at -20°C. The cell pellets were lysed with Radioimmunoprecipitation Assays buffer (RIPA: 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% Triton X-100 and 0.1% SDS) and the cOmplete™ Mini protease inhibitor-cocktail (Merck, Darmstadt, Germany) for 30 min on ice. Samples were subsequently centrifuged at 11,000 g for 10 min at 4°C. The ensuing cell lysate supernatants were transferred into a fresh tube and stored at -20°C until further use.
Protein concentrations were determined with a Bradford assay (Roti®-Nanoquant #K880.1, Carl Roth, Karlsruhe, Germany) according to the manufacturer’s protocol. The measurement was performed on a Fluostar Omega (BMG Labtech, Ortenberg, Germany) at 590/450 nm.
SDS-PAGE was performed with 40 µg protein from cell lysates and 5 µg protein from supernatant reduced in 4 × Laemmli-buffer (Roti®-Load1, #K929.1, Carl Roth, Karlsruhe, Germany). Samples were denaturized at 95°C for 5 min and subsequently loaded onto 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad, Hercules, CA, USA). Protein bands were seperated at 100 V for 1 h with Western Blot Running Buffer (25 mM TRIS Base, 192 mM Glycin, 0.1% SDS; pH 8.3) using the Mini-PROTEAN® Tetra Vertical Electrophoresis System (Bio-Rad, Hercules, CA, USA). After separation, gels were blotted onto a nitrocellulose membrane (#GE10600003, Amersham™ Protran® Premium, Sigma-Aldrich, Taufkirchen, Germany) at 100 V for 1 h (Mini Trans-Blot Module, Bio-Rad, Hercules, CA, USA).
Membranes were washed in Tris-buffered saline with Tween solution (TBST: 20 mM TRIS, pH 7.5, 150 mM NaCl, 0.1% Tween-20) and blocked with TBST containing 5% milk powder (Carl Roth, Karlsruhe, Germany) for 1 h at room temperature. Immunostaining of GFP fusion proteins of cell lysate and supernatant samples was done with GFP Polyclonal Antibody, DyLight™ 800 (1:10,000 dilution; #600–145-215, Thermo Fisher Scientific, Waltham, MA, USA). HA tagged proteins within cell lysate and supernatant samples were immunostained with HA Tag Monoclonal Antibody (2–2.2.14), DyLight™ 550 (1:1000 dilution; #26183D550, Thermo Fisher Scientific, Waltham, MA, USA). GAPDH Loading Control Monoclonal Antibody (GA1R), DyLight™ 680, Thermo Fisher Scientific, Waltham, MA, USA, was used to normalize the Western blot. The blots were stained at 4°C overnight and washed the next day three times for 10 min in TBST. A ChemiDoc Imaging System, Bio-Rad (Hercules, CA, USA) was used for imaging.
GAPDH loading control monoclonal antibody was used for normalization of protein amount from cell lysates. Supernatants were normalized to the total protein content within the stain-free gels. A ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA) was used for imaging.
Statistical analysis
Basic statistics (mean values and standard deviations) were performed using Microsoft Excel 2016 (Office Professional Plus 2016; Microsoft, Redmond, WA, USA) and visualized using GraphPad Prism (version 9.51). Results are shown as mean values ± standard deviation. One-way ANOVA followed by Bonferroni and Holm multiple comparison (all pairs simultaneously compared) (https://astatsa.com/) was used to test for differences between cell lines.
In silico prediction of protein structure
Tertiary structure of WT and mutated AT was predicted using Contact-guided Iterative Threading ASSEmbly Refinement (C-I-TASSER) [9]. Furthermore, iCn3D was used to display the calculated models [10, 11].


Comments (0)