
Remember me
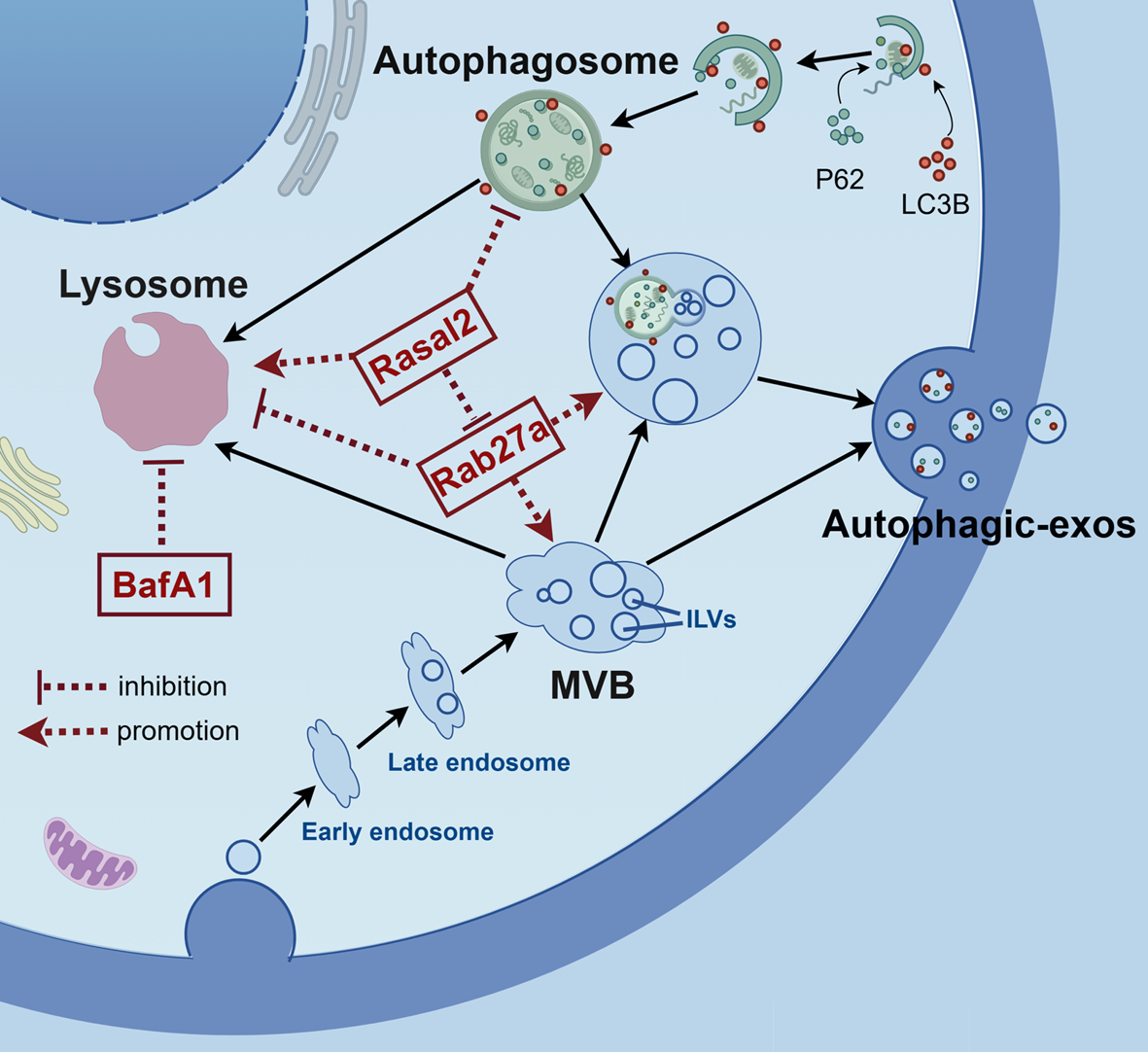
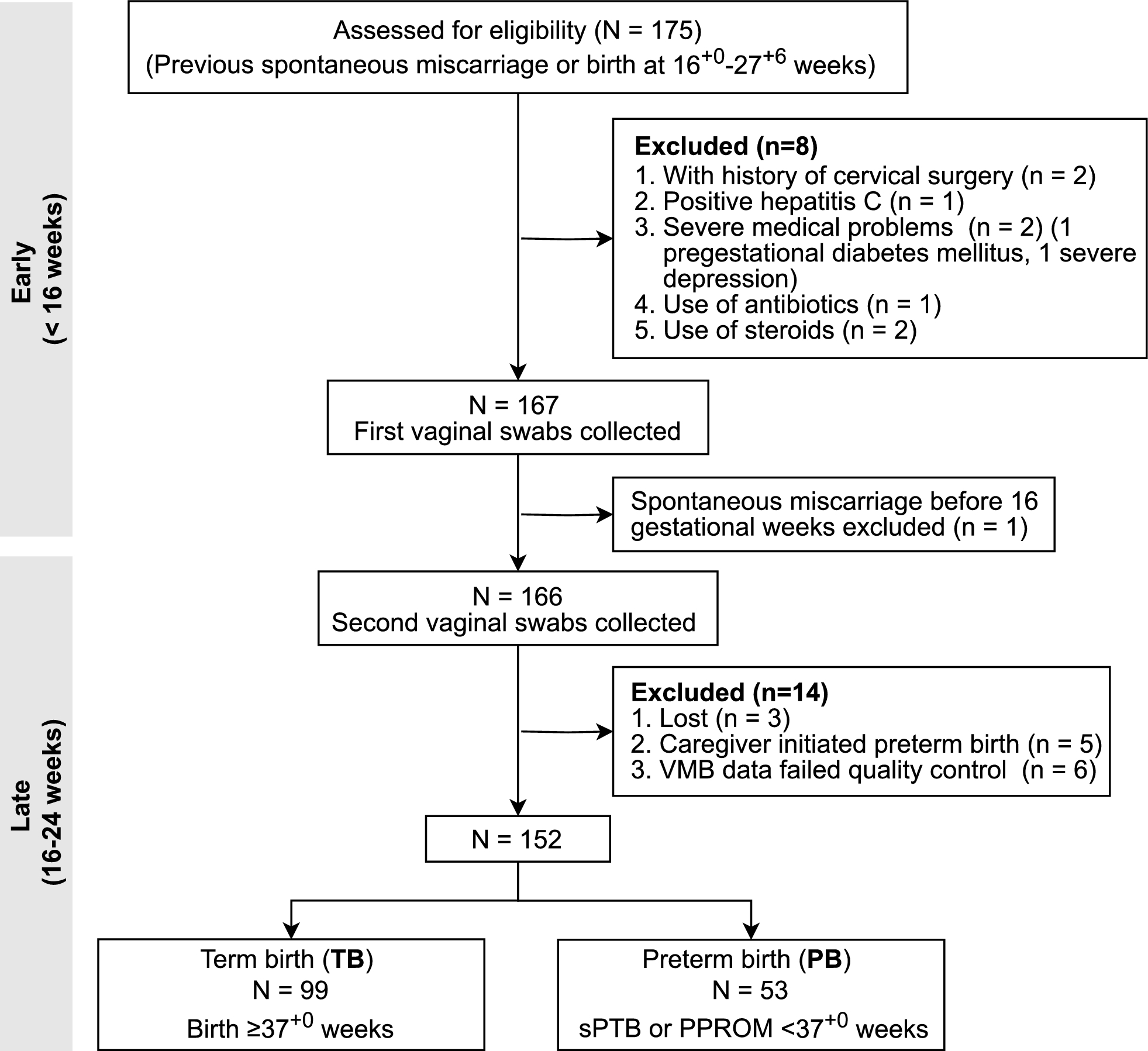




The data was mined and harmonized from more than 30 sources and the data architecture was designed in the advanced MVVM framework with separated layers for data access on multiple levels. A reporting system was also developed in the backend for automated annotation of external variant data, enabling scalable and portable implementation of patient data interpretation (Fig. 1).
Fig. 1
An overview of the flowchart of data construction of Tri©DB and architecture of the platform. Top: illustration of database elements and contents; Middle: key data processing procedures; Bottom: the layout and key techniques in the architecture of Tri©DB
The database comprises 398,180 population-level alterations on 1,308 altered genes, 232 actionable genotypes, 84 cancer types, 268 therapies linked by 948 associations, 1,847 clinical trials, and 40 annotation entities by mining more than 33 external databases and numerous literatures (see “Methods and Materials” section). Key statistic summary of genes, diseases, and therapies is presented in Fig. 2. It is found that the majority of the alterations are missense mutations, and KMT2D contains the most mutated loci, followed by APC, ARID1A, and TP53 (Fig. 2A). Up to now, combination products are the major form of cancer therapies and Genentech developed the most new molecular drugs, followed by Novartis and Pfizer (Fig. 2B). For the cancer types mentioned in Tri©DB, a clickable circle tree was used to demonstrate their classification in a hierarchical structure (Fig. 2C). We further assessed the profile of gene mutation prevalence in the major cancer types (Fig. 2D and E). Several of the hotspot genes have been successful targets for the development of anticancer drugs, such as BRAF, EGFR, and KDR, while the majority of commonly altered genes have no drugs available.
Fig. 2
Graphical presentation of the key statistics of data included in Tri©DB. A The gene mutation classifications (inner circle) and the top genes with the most mutated loci (outer circle). B The drug classifications (inner circle) and top manufactures (outer circle). C The hierarchical classification of the cancer types mentioned in Tri©DB. Each dot can be clicked to show the subtypes of each main cancer type. D Heatmap presentation of the prevalence of gene alterations in the major cancer types listed in Tri©DB based on the GENIE cohort. E The same as that in C but only for alterations annotated as pathogenic/likely pathogenic. Only the most commonly altered or well-known cancer driver genes are shown. The existence of drugs for the altered genes is indicated in coloured bars on top of the heatmap
Derivation of data in gene-, disease-, and therapy-oriented tabular format in the first layer of dataThe content of the knowledgebase can be accessed in two layers from the web interface to accommodate the needs of diverse user groups. The first layer provides all gene-disease-therapy triple-relationships along with fifteen annotation attributes in a brief tabular format in the gene-, disease-, and therapy-oriented view. The second layer presents the detailed annotation and interpretation in a separate page for each entity.
The tabular data presentation in the first layer aims to provide an overview yet with sufficient annotation of the gene-disease-therapy triple-relationships. The data can be obtained in the gene-, disease-, therapy-oriented view separately. The three oriented views contain various annotation information (Table 1).
Table 1 The annotatiaon attributes in the gene-, disease-, and therapy-oriented tabular viewsThe implementation of the three separate views was done by recognizing the complexity of the relationship between gene alterations, disease phenotypes and therapies, where an individual alteration might occur in different disease contexts and are predictive of responses to distinct therapeutic interventions, and vice versa, an individual disease could be related with multiple gene alterations involved in different biological pathways and call for differentiated therapy protocols. The design will facilitate rapid access to key information for users from diverse background without digging into additional details, and the data can be seamlessly integrated into third-party reporting systems or annotation pipelines.
Unique features provided by the first layer of dataIn the first layer of the data, our database provides two unique features, which have not been present in other similar resources. Firstly, in addition to the regular positive genotypes (gene + alteration), we added the attribute “Negative Genotypes” to indicate the opposing genotypes, which may not respond to a specified therapy or is associated with poor prognosis. For example, the recent anti-EGFR therapies for metastatic colorectal carcinoma should not be used in patients with KRAS mutations [46].
Secondly, we identified “Direct Target” in the therapy-oriented tabular view for each therapy for which the matching genotypes are different from the directly targeted genes. A notable example is the therapies for KRAS mutated carcinomas. Before the successful development of KRAS inhibitor Sotorasib, the therapeutic studies for KRAS-mutant cancers focused on targeting downstream effectors in the RAS-RAF-MEK-ERK pathway, such as the MEK inhibitor Trametinib in combination with chemotherapy for patients with metastatic non-small cell lung cancer (NSCLC) [47].
Derivation of detailed annotation information for each gene-disease-therapy triple-relation in the second layer of dataThe second layer of data in Tri©DB aims to provide detailed annotation for each record of gene-disease-therapy triple-relationships, offering a rich breath of cancer-related knowledge in structured attributes, such as functional annotations, interpretations, population carrier rate, and interactive networks.
As an example, the details for the gene EGFR (epidermal growth factor receptor), therapeutic drug osimertinib, and disease NSCLC was illustrated in Fig. 3. For the gene EGFR, the results show that 98 different alterations occurring among the GENIE cancer cohort, whereas the most common alterations are exon 21 missense mutations, exon 19 deletion mutations, and exon 20 mutation T790M (the lollipop graph in Fig. 3A). To provide mechanistic explanations for the pathogenesis of the genes and the therapies relevant to the specific gene, the recapitulative interpretations for each gene, i.e., “Functional and Clinical Implications” and “Clinical Interpretations” were constructed based on intensive literature review and manual curation. In complement with the text interpretation, a graphical presentation “Pathway and Interaction” was provided by connecting to three external resources, i.e., REACOME [33], KEGG [32] and NCG [34].
Fig. 3
An example of the detailed report for the gene EGFR, therapeutic drug osimertinib, and disease NSCLC in the second layer of data in Tri©DB. A Details of EGFR, including basic genomic annotation, population mutation profile, functional interpretation, therapy interpretation, pathway and interaction. B Details of the drug osimertinib, including approval information, indication, mechanism of action, clinical trial, and the three-dimensional structure. C Details of NSCLC, including disease classification, mutation carrier rate in the Western and Chinese population. D Germline mutation carrier rate was also provided in the disease report
The small molecular inhibitor osimertinib is the third-generation TKI to overcome resistance mediated by EGFR mutations including T790M. The report for this drug presents multi-dimensional information on the mechanistic and clinical level, such as the three-dimensional complex structure of osimertinib and EGFR-T790M, and the clinical trials for validating efficacies of osimertinib (Fig. 3B). This information might be of particular interest for health care professionals or patient groups who are seeking to enroll in trials relevant to a specific drug.
The cancer-type level calculation of the population carrier rate of gene alterations among the NSCLC cohort shows that KRAS mutations are the most common somatic alterations in the Western population of the NSCLC (> 20%), while EGFR variations account for the largest proportion in the Chinese population (Fig. 3C). The results of the population carrier rate for the cancer cohort provide a brief idea of the fraction of patient population who may benefit from the therapies targeting a specific gene.
Unique features of the detailed annotation information in the second layer of dataTri©DB provides multiple unique features for the detailed annotation in the second layer of data. The most notable includes the following three. First, Tri©DB collected and compiled the cancer-level population carrier rate for germline mutations (Fig. 3D). This information was generally ignored by other resources probably due to the overall low population prevalence [25]. For example, the germline mutations are rare in NSCLC with the most common mutation occurring in the homologous recombination repair (HRR) gene CHEK2 (S471F, 0.295%) (Fig. 3D). The top germline mutations were also found in several other HRR genes, such as ATM (V2716A, 0.197%), FANCC (R185*, 0.098%), BRCA2 (R3128*, 0.098%). (Fig. 3D).
Secondly, Tri©DB constructed the disease-gene-therapy triple-relationships in a disease-centred manner and dynamically generated interactive networks for the triple-relations (Fig. 4). The network presentation will help to elucidate the genetic and therapeutic landscape for a specific cancer type. Users can interact with the networks by refining the layout or redirecting to internal and external resources for further details of each node in the network. An example of the landscape for the colorectal cancer is demonstrated in Fig. 4. It shows that more than 10 altered genotypes, such as TP53, APC, KRAS, and BRAF, and the global DNA instability (i.e., Microsatellite Instability High or Mismatch repair deficiency, namely MSI-H/dMMR) have been found to be associated with colorectal cancer. Nine of them have approved therapies to act on their altered form, such as BRAF, ERBB2, KRAS, EGFR, VEGFA, VEGFR/KDR, FLT1, FLT4, and MSI/MMR.
Fig. 4
Notable features of the detailed annotation information for the example disease colorectal cancer. The interactive network presentation of the gene-disease-therapy triple-relationship with the colorectal cancer as the network centre. The sizes of gene nodes and the weights of disease-gene edges are proportional to the accumulated carrier rate of gene-level alterations in cancer-specific population cohort. Each node of genes or therapies is linked to multiple external resources
Thirdly, our database presents the mechanism-based cancer-specific pathways, which have been largely scattered around literatures or databases. We collected and mined those pathways by thorough literature survey. The links of the source of origin are also provided. An example is the pathway map for colorectal cancer [48]. It is shown that colorectal cancer can develop via multiple genetic (APC, KRAS, TP53, BRAF, MMR) and epigenetic (MLH1) factors involving several distinct but intertwined pathways, such as Wnt signalling pathway, Myc signalling pathway, MAPK pathway, TGF-β pathway, and serrated neoplasia pathway. The mechanism pathways of cancer types in combination with the disease-gene-therapy networks provide valuable pivot points for elucidating the pathogenic and therapeutic landscape of specific cancers.
Automatic annotation and generation of the portable interpretation reportIn addition to the interactive access to the data in Tri©DB, our open-source platform also contains web interface reporting system facilitating automated annotation and interpretation of user-provided bulk variant data.
The reporting system supports a variety of variant classes (including SNV, CNV, SV, MSI, somatic mutations, and germline mutations) for uploading in standard or software-specific formats. The system also allows users to designate the mutation types (i.e., somatic or germline) and sequencing modalities (i.e., WGS, WES, or Gene Panel) for adapting to distinct analysis workflow or knowledgebase contents (Fig. 5A). Considering the diverse types of genomic alterations relevant to cancer, the annotation system at first performs multiple analysis, such as tumor mutation burden (TMB) calculation, MMR gene detection, HRR gene detection, mutational signature identification, and subsequently matches each variant signature against Tri©DB for extracting multiple annotations, such as gene functions relevant to cancer, clinical trials, mechanism of actions, drug resistance, and et al. Finally, all the analysis and annotation results are organized and integrated in a single report file enabling easy dissemination and communication among researchers (Fig. 5B and Additional file 1).
Fig. 5
Overview of the reporting system. A The web interface of the file uploading module allowing users to provide various mutation types and sequencing modalities. B Preview of the integrated report for user-input variant data
Performance evaluation of Tri©DB in variant annotationTo evaluate the performance of Tri©DB in variant annotation, we made therapy matching using Tri©DB in two scenarios, i.e., an individual patient sample and a patient cohort and compared the matching with that from other resources.
Firstly, an artificial individual patient was created to harbor 37 variants from 10 genes representing a wide range of variant types (SNV, gene amplification, gene loss, and fusion) and driver gene categories (such as cell proliferation, apoptosis inhibition, angiogenesis, DNA repair, and genomic instability) (Additional file 2). The variants were annotated using Tri©DB and compared the annotation with that using two notable knowledgebases of similar kind, i.e. oncoKB which was recognized by FDA to support cancer precision medicine practices and MCG (My Cancer Genome) which was commercially licensed. Based on FDA evidences and clinical guidelines, a total of 420 treatment options for the 37 variants (i.e. variant-disease-therapy triples) were annotated by Tri©DB sharing 97.9% of those by oncoKB and 99.6% of those by MCG. Tri©DB annotated 133 more treatments than oncoKB and 137 than MCG, accounting for 46% of the shared list (Additional file 2). The additional treatments annotated by Tri©DB are mainly targeted antibody drugs or immunotherapy drugs, such as necitumumab and durvalumab for EGFR, and pertuzumab for ERBB2.
We then made therapy matching for a patient cohort from a prospective clinical trial called I-PREDICT dedicated to investigate individualized cancer therapy (NCT02534675) [49]. This clinical trial administered individualized therapies for 83 patients diagnosed of a wide range of cancer types and has been used for therapy recommendations by MOAlmanac, an integrative platform of clinical interpretation [50]. A total of 524 gene variants from the 83 patients were extracted and therapy matching was made by Tri©DB on the per-variant per-patient basis. Based on FDA evidences or clinical guidelines, 59 variant-patient pairs were administered with therapies in the clinical trials, 56 of which (94.9%) are overlapped by Tri©DB involving 31 patients (Additional file 3). The three annotations missed by Tri©DB are all tamoxifen, which was approved by FDA 40 years ago. The overlap proportion is significantly higher than that for MOAlmanac (20 variant-patient pairs, 33.8%), probably because MOAlmanac focused on providing best therapy recommendations but not a comprehensive list based on the global molecular profile of each patient.
The comparison results highlight the high completeness and accuracy of the annotations by Tri©DB. The high consistency of Tri©DB with the clinical trial practices supports its utility in clinical applications.
Comments (0)