
Remember me
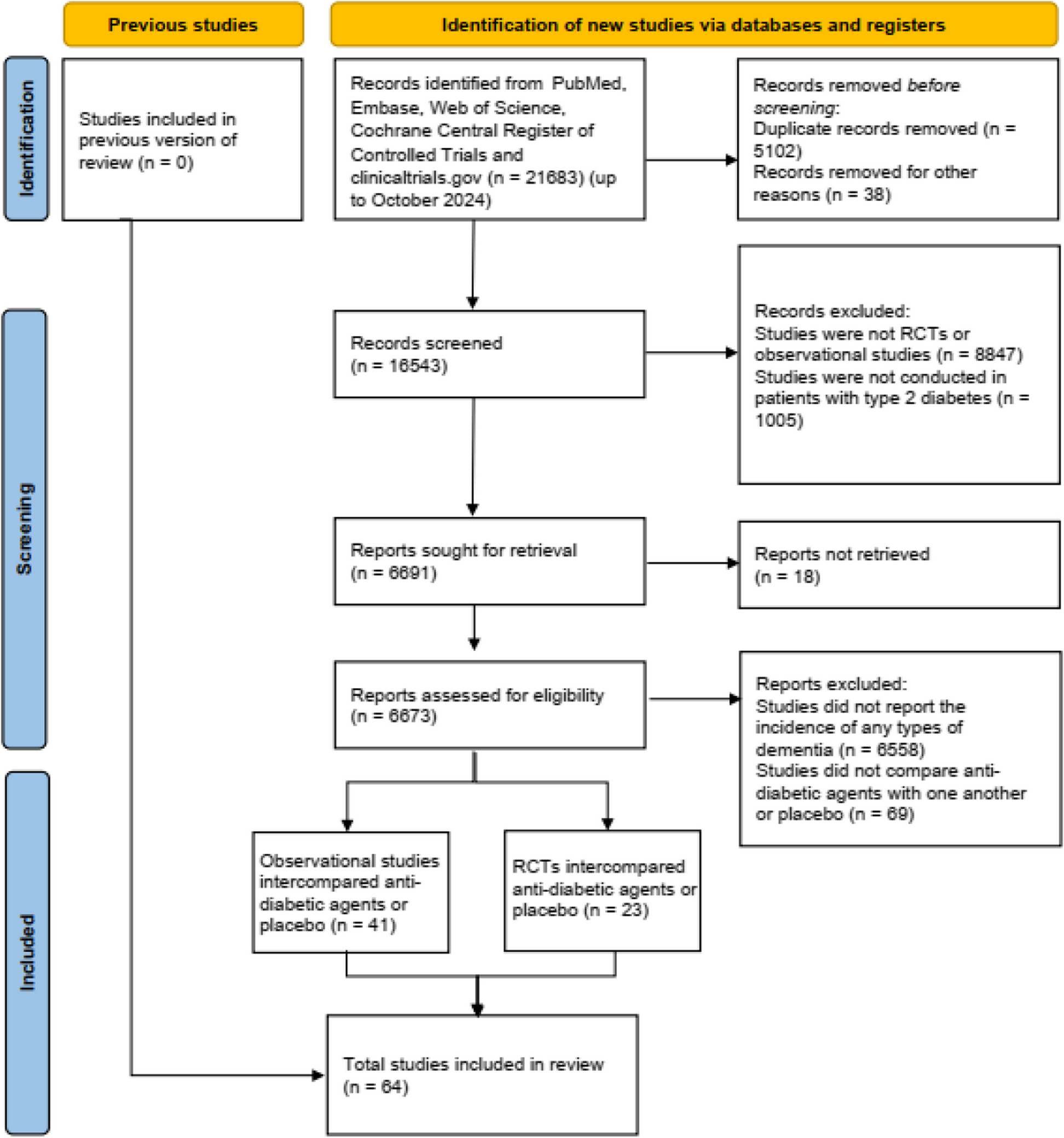
Figure 1 presents an overview of our overall analysis strategy, and the details can be found in the “Methods” section. We make use of MRI and positron emission tomography (PET) from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). In summary, we use diffusion MRI to generate the structural connectomes of healthy controls (HC), mild cognitive impairment (MCI), and Alzheimer’s disease (AD) subjects. We use task-free resting-state functional MRI to fit a whole-brain model in which the local neuronal dynamics of each brain region evolve according to the dynamic mean field model by Deco et al. [18], which is then connected to a spontaneous blood-oxygenation-level-dependent (BOLD) dynamics. We refer to this model as the balanced excitation-inhibition (BEI) model, which can be thought of as a homogeneous reference against which we evaluate the performance of our heterogeneous AD model. A\(\beta\) and tau distributions are derived from AV-45 and AV-1451 PET from ADNI. For the heterogeneous model, we incorporate regional heterogeneous distributions of the main proteins involved in AD, namely A\(\beta\) and tau, as first-order multiplicative polynomials for each burden and for each type of population (excitatory/inhibitory) into the local gain parameter \(M_\). Fitting the model to empirical fMRI data allows us to evaluate which effect of A\(\beta\) and tau on the different populations can mechanistically explain the observed behaviors.
Fig. 1
Illustrative overview of our processing pipeline. A Basic ingredients for the integration of protein burden data from structural (dMRI, top left), functional (fMRI, top right), and burden (PET, right) using the same parcellation for each neuroimaging modality (top, middle) for generating a whole-brain computational model (bottom left). Each node of the model is using a realistic underlying biophysical neuronal model including AMPA (blue connections), GABA (red), and NMDA (gray) synapses as well as neurotransmitter gain modulation of these. B Fitting the measures in the whole-brain model: First, we simulate the BOLD timeseries for each brain region in the parcellation, for each subject. These timeseries are defined by its inputs, namely a previously found global coupling constant G, an individual Structural Connectivity (SC) matrix, and the corresponding individual A\(\beta\) and tau burdens. Subsequently, we compute a time-versus-time matrix of phase functional connectivity dynamics (phFCD). This is compared to a reference empirical phFCD extracted from the fMRI data off the same subject using the Kolmogorov-Smirnov distance (KS), \(D_\), which is minimized to find the generative parameters of the model. This process is repeated for the other two measures of brain dynamics, functional connectivity (FC) and sliding-window functional connectivity dynamics (swFCD)
Model fittingFor both of our models, homogeneous and heterogeneous, we assume that all diffusion MRI-reconstructed streamline fibers have the same conductivity, and thus the coupling between different brain areas is scaled by a single global parameter, G. We first tune the G parameter of the BEI model to adjust the strength of effective coupling in the model and identify the brain’s dynamic working point by fitting the model to three empirical properties that are estimated from the empirical fMRI data:
The Pearson correlation between model and empirical estimates of static (i.e., time-averaged) functional connectivity estimated across all pairs of brain regions (FC);
Similarity in sliding-window functional connectivity dynamics (swFCD); and
The KS distance between a set of dynamic functional connectivity matrices (also called coherence connectivity matrix [21]) built from the average BOLD time series of each ROI, which were Hilbert-transformed to yield the phase evolution of the regional signals (phFCD).
We then fit the coefficients for the two burdens, for excitatory and inhibitory populations, with a global optimization algorithm, within directional bounds obtained from previous clinical observations (see below, in the “Constraints” section).
Result evaluationTo demonstrate that E/I imbalance is dependent on the precise distribution of the A\(\beta\) and tau burdens, at the optimal values obtained with the fitting procedure described above, we randomly shuffled the empirical protein burdens; i.e., the original 378 values for each of the misfolded protein maps were randomly re-assigned to different regions, and the model was run 10 times with each different randomly re-assigned receptor map, and the simulation was repeated 10 more times for each re-assigned receptor map, for a total of 100 simulations each time. Figure 5 shows the results of randomly shuffling the empirical burden densities across the regions at the optimum point. This randomly reshuffled manipulation yields a significantly worse fit compared to the actual empirical burden densities (as shown by the Wilcoxon statistics in the boxplot). We additionally evaluate the quality of the simulation results with the optimized parameters with original (i.e., not shuffled) burdens and with the homogeneous BEI model. Finally, we examine the relevance of each type of burden by optimizing them in isolation from each other (i.e., zeroing the other one out), and comparing the results. The whole comparisons include both burdens in isolation, both burdens simultaneously, and with the homogeneous (i.e., BEI) model.
ParticipantsEmpirical data were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu), which is a longitudinal multi-site study designed to develop biomarkers for Alzheimer’s disease (AD) across all stages. The inclusion criteria for AD patients was the NINCDS-ADRDA criteria, which contains only clinical features [22], and an MMSE score below 24. For both HC and MCI, the inclusion criteria were a MMSE (Mini-Mental State Examination) score between 24 and 30, as well as age between 55 and 90 years. Also, for MCI, participants had to have a subjective memory complaint and abnormal results in another neuropsychological memory test. Imaging and biomarkers were not used for the diagnosis.
Data acquisition and processingAll the data in this study were previously used in Stefanovski et al. [15] work, so we will present here an abridged version of the processing performed on the original data and refer to the original work for the details. All images used in this study were taken from ADNI-3, using data from Siemens scanners with a magnetic field strength of 3T.
Structural MRIFor each included participant, we created a brain parcellation for our structural data using FLAIR, following the minimal preprocessing pipeline [23] of the Human Connectome Project (HCP) using FreesurferFootnote 1 [24], FSL [25,26,27], and connectome workbenchFootnote 2. Therefore, we used T1 MPRAGE, FLAIR, and fieldmaps for the anatomical parcellation. We then registered the subject cortical surfaces to the parcellation of Glasser et al. [28] using the multimodal surface matching (MSM) tool [29]. In this parcellation, there were 379 regions: 180 left and 180 right cortical regions, 9 left and 9 right subcortical regions, and 1 brainstem region.
PET imagesFor A\(\beta\), we used the version of AV-45 PET already preprocessed by ADNI, using a standard image with a resolution of 1.5mm cubic voxels and matrix size of \(160 \times 160 \times 96\), normalized so that the average voxel intensity was 1 and smoothed out using a scanner-specific filter function. Then, a brainmask was generated from the structural preprocessing pipeline (HCP) and used to mask the PET image. We received in each voxel a relative A\(\beta\) burden which is aggregated according to the parcellation used for our modeling approach. Subcortical region PET loads were defined as the average SUVR in subcortical gray matter (GM), normalized by the intensity of the cerebellum. With the help of the connectome workbench tool, using the pial and white matter surfaces as ribbon constraints, we mapped the Cortical GM PET intensities onto individual cortical surfaces. Finally, using the multimodal Glasser parcellation we derived average regional PET loads.
For tau, we also used ADNI’s preprocessed version of AV-1451 (Flortaucipir) following the same acquisition and processing, resulting in a single relative tau value for each voxel. Then, these values were also aggregated to the selected parcellation, following the already mentioned steps. The final average regional tau loads were obtained in the Glasser parcellation. Both mean burden values can be found, for each cohort, in Table 2.
DWIIndividual tractographies were computed only for included HC participants, and they were averaged to a standard brain template (see below). Preprocessing was mainly done with the MRtrix3 software packageFootnote 3.
In particular, the following steps were performed: First, we denoised the DWI data [30], followed by motion and eddy current correctionFootnote 4. Then, B1 field inhomogeneity correction (ANTS N4), followed by a brainmask estimation from the DWI images. Next, we normalized the DWI intensity for the group of participants, which was used to generate a WM response function [31], and created an average response function from all participants. Next, we estimated the fiber orientation distribution and the average response function [32] using the subject-normalized DWI image, to generate a five-tissue type image. Finally, we used the iFOD2 algorithm [33] and the SIFT2 algorithm [34] to get the weighted anatomical constrained tractography [35], to end up merging all information into the Glasser connectome, resulting in a structural connectome (SC).
The participating centers of ADNI are following centrally controlled protocols, which are described in detail at the ADNI websiteFootnote 5. This includes the set-up of the MRI protocol by central ADNI institutions and quality checks with phantoms. However, there indeed remain differences between ADNI centers and ADNI phases. In the Supplemental material, we added a table with the DTI metadata of the 15 healthy controls that were used for structural connectivity calculation in this study (from [15]). Here, we see, despite the focus on only one scanner type from Siemens, that the data uses two different MRI protocols that only slightly differ: ADNI3 basic and ADNI3 advanced, which were designed to be compatible with each otherFootnote 6. The main difference remains in the TR and TE values, due to the different extent of measured sequences. A recent study showed for ADNI data including the used protocols that a cross-center harmonization indeed leads to significant differences in diffusion imaging derivatives, but the relationship with clinical data did not change, no matter if the harmonization was performed or not [36]. We, therefore, employed standardized preprocessing steps including quality controls, as outlined in the minimal processing pipeline of the Human Connectome Project [23]. To address the remaining effects, we have employed a methodological approach to compensate for this bias: we decided to use a group-based structural connectivity template for all participants. This comes with the benefit of standardizing the underlying network to an unaffected brain, which is then influenced by A\(\beta\) and Tau. Even though the group-based template is derived from data from several ADNI study sites, this approach protects from introducing artificial differences between the subjects, as all are using the same structural connectivity. Also, it must be taken into account that we used an averaged SC from all healthy controls, thus further reducing any systematic bias of this kind.
fMRIWith respect to the processing of the fMRI data, the images were initially preprocessed in FSL FEAT and independent component analysis-based denoising (FSLFIX) following a basic pipeline [15]. Time courses for noise-labeled components, along with 24 head motion parameters, were then removed from the voxel-wise fMRI time series using ordinary least squares regression.
The resulting denoised functional data were spatially normalized to the MNI space using Advanced Normalization Tools (version 2.2.0). Mean time series for each parcellated region were then extracted, and interregional FC matrices were estimated using Pearson correlations between each pair of regional time series. Dynamic FC matrices were also calculated for the empirical data, as outlined below.
Generation of a standard brain templateAs previously done [15], we average the SCs of all HC participants, using an arithmetic mean
$$\begin C_\mu = \left( \sum \limits _^n C_i\right) /n = (C_1 + C_2 + . . . + C_n)/n \end$$
wherein \(C_\mu\) is the averaged SC matrix, n is the number of HC participants and \(C_i\) is the individual SC matrix.
However, as matrices in this context are large (i.e., 379 regions), the average input to any given node can be too large for the DMF, making fitting and processing in general more difficult. Thus, we discarded the traditional normalization of dividing the matrix elements by their maximum, and used a slightly different approach, instead. First, we added one and applied the logarithm to every entry, as \(lC = log(C_\mu +1)\). Then, we computed the maximum input any node could receive for a unitary unit input current, \(maxNodeInput = max_j(\sum _i(lC_))\), and finally, we normalized by \(0.7 * lC / maxNodeInput\), where 0.7 was chosen to be a convenient normalization value. Observe that this constant is actually multiplying another constant G in the model which we fit to empirical data, so its actual value can safely be changed.
In Fig. 3, we can find the SC matrix and organization graph, where we can observe that the general characteristics of physiological SCs such as symmetry, laterality, homology, and subcortical hubs are maintained in the averaged connectome. The election of the averaged SC allowed us to control all factors (e.g., atrophy), which matched our objective of simulating the activity from both healthy and “pathogenic” modifications by A\(\beta\) and tau.
Balanced excitation-inhibition (BEI) modelIn this work, we used the dynamic mean field (DMF) model proposed by Deco et al. [18], which consists of a network model to simulate spontaneous brain activity at the whole-brain level. Following the original formulation, each node represents a region of interest (i.e., a brain area) and the links represent the white matter connections between them. In turn, each node is a reduced representation of large ensembles of interconnected excitatory and inhibitory integrate-and-fire spiking neurons (as in the original, respectively 80% and 20% neurons), to a set of dynamical equations describing the activity of coupled excitatory (E) and inhibitory (I) pools of neurons, based on the original reduction of Wong and Wang [37]. In the DMF model, excitatory synaptic currents, I(E), are mediated by NMDA receptors, while inhibitory currents, I(I), are mediated by \(GABA_A\) receptors. Both neuronal pools are reciprocally connected, and the inter-area interactions occur at the excitatory level only, scaled by the structural connectivity \(C_\) (see the “Structural MRI” section).
To be more specific, the DMF model is expressed by the following system of coupled differential equations:
$$\begin I_k^ = W_E\,I_o + w_+\,J_N \, S_k^ + J_N G \sum \limits _j C_ S_j^ - J_k S_k^+ I_ \end$$
(1)
$$\begin I_k^ = W_I\,I_o + J_N S_k^ - S_k^ + \lambda J_N G \sum \limits _j C_ S_j^ \end$$
(2)
$$\begin r_k^ = H^\left( I_k^\right) = \frac - b_E\right) }\left( -d_E M_k^E \left( a_E I_k^ -b_E\right) \right) } \end$$
(3)
$$\begin r_k^ = H^\left( I_k^\right) = \frac - b_I\right) }\left( -d_I M_k^I \left( a_I I_k^ -b_I\right) \right) } \end$$
(4)
$$\begin \dot_k^ = -\frac} + \left( 1 - S_k^\right) \, \gamma H^\left( I_k^\right) \end$$
(5)
$$\begin \dot_k^ = -\frac} + H^\left( I_k^\right) \end$$
(6)
Here, the last two equations should add, when integrating, an uncorrelated standard Gaussian noise term with an amplitude of \(\sigma = 0.01nA\) (using Euler-Maruyama integration). In these equations, \(\lambda\) is a parameter that can be equal to 1 or 0, indicating whether long-range feedforward inhibition is considered (\(\lambda = 1\)) or not (\(\lambda = 0\)). In the above equation, the kinetic parameters are \(\gamma = 0.641/1000\) (the factor 1000 is for expressing everything in ms), \(\tau _E = \tau _\), and \(\tau _I = \tau _\). The excitatory synaptic coupling \(J_ = 0.15\) (nA). The overall effective external input is \(I_0 = 0.382\) (nA) scaled by \(W_E\) and \(W_I\), for the excitatory pools and the inhibitory pools, respectively. The effective time constant of NMDA is \(\tau _= 100\) ms [37]. The values of \(W_I\), \(I_0\), and \(J_\) were chosen to obtain a low level of spontaneous activity for the isolated local area model. The values of the gating variables can be found in Table 1.
Table 1 Gating variables in the BEI modelAs mentioned, the DMF model is derived from the original Wong and Wang model [37] to emulate resting-state conditions, such that each isolated node displays the typical noisy spontaneous activity with low firing rate (\(H^\sim 3Hz\)) observed in electrophysiology experiments, reusing most of the parameter values defined there. We also implemented the feedback inhibition control (FIC) mechanism described by Deco et al. [18], where the inhibition weight, \(J_n\), and was adjusted separately for each node n such that the firing rate of the excitatory pools \(H^\) remains clamped at 3 Hz even when receiving excitatory input from connected areas. Deco et al. [18] demonstrated that this mechanism leads to a better prediction of the resting-state FC and to a more realistic evoked activity. We refer to this model as the balanced excitation-inhibition (BEI) model. Although the local adjustments in this model introduce some degree of regional heterogeneity, the firing rates are constrained to be uniform across regions so we consider this BEI model as a homogeneous benchmark against which we evaluate more sophisticated models that allow A\(\beta\) and tau to affect intrinsic dynamical properties across regions.
Following the Glasser parcellation [23], we considered \(N = 379\) brain areas in our whole-brain network model. Each area n receives excitatory input from all structurally connected areas into its excitatory pool, weighted by the connectivity matrix, obtained from dMRI (see the “DWI” section). Furthermore, all inter-area E-to-E connections are equally scaled by a global coupling factor G. This global scaling factor is the only control parameter that is adjusted to move the system to its optimal working point, where the simulated activity maximally fits the empirical resting-state activity of healthy control participants. Simulations were run for a range of G between 0 and 5.5 with an increment of 0.05 and with a time step of 1 ms. For each G, we ran 200 simulations of 435 s each, in order to emulate the empirical resting-state scans from 17 participants. The optimum value found, for the phFCD observable, is for \(G=3.1\). See Fig. 2A.
Fig. 2
Optimization and evaluation of the model: First, using only HC subjects, the global coupling parameter G is found, and then the model is adjusted to minimize the distance between the empirical and simulated fMRI data, taking into account the regional burden distributions. A Minimization of G between 0 and 5.5, for functional connectivity (FC), sliding-window functional connectivity dynamics (swFCD), and phase FCD (phFCD). Given their strong similarity in the results, phFCD was used for all subsequent computations. B, C Shows the normalized (in [0, 1]) FCD distributions for the empirical data (top) and the simulated model at the optimal result (bottom). D, E, F Analysis of the impact (smaller values are better) of the different burdens with respect to their impact on the phFCD (KS distance) when optimized together and in isolation, with the homogeneous state as a reference. Clearly, in all cases, the combined burden outperforms any other model. However, as can be seen, the results for AD clearly show that tau alone accounts for the vast majority of the weight of the impact on brain activity (F), while for MCI patients it is A\(\beta\) who dominates (E). For HC patients we also see a predominance of A\(\beta\), although with less difference between the model incorporating A\(\beta\) and tau vs. A\(\beta\) in isolation (D). Average distributions of A\(\beta\) (G) and tau burdens (H) over each cohort (using ADNI’s database). Colors correspond to the normalized burden of each protein. The increase in A\(\beta\) and tau can be clearly seen
Simulated BOLD signalOnce we have obtained the simulated mean field activity, we need to transform it into a BOLD signal we used the generalized hemodynamic model of Stephan et al. [38]. We compute the BOLD signal in the k-th brain area from the firing rate of the excitatory pools \(H^\), such that an increase in the firing rate causes an increase in a vasodilatory signal, \(s_k\), that is subject to auto-regulatory feedback. Blood inflow \(f_k\) responds in proportion to this signal inducing changes in blood volume \(v_k\) and deoxyhemoglobin content \(q_k\). The equations relating these biophysical variables are:
$$\begin \frac & = 0.5 r_k^ + 3 - k s_k - \gamma (f_k-1) \nonumber \\ \frac & = s_k \nonumber \\ \tau \frac & = f_k - v_k^} \\ \tau \frac & = f_k \frac}} - q_k \frac}}\nonumber \end$$
(7)
with finally
$$\begin B_k = v_0 \left[ k_1(1-q_k)+k_2\left( 1-\frac\right) +k_3(1-v_k) \right] \end$$
being the final measured BOLD signal.
We actually used the updated version described later on [38], which consists on introducing the change of variables \(\hat = ln z\), which induces the following change for \(z=f_k\), \(v_k\) and \(q_k\), with its corresponding state equation \(\frac = F(z)\), as:
$$\begin \frac} = \frac \frac = \frac \end$$
which results in \(z(t)=exp(\hat(t))\) always being positive, which guarantees a proper support for these non-negative states, and thus numerical stability when evaluating the state equations during evaluation.
A\(\beta\)-Tau modelIn our heterogeneous model, A\(\beta\) and Tau are introduced, at the formulae for the neuronal response functions, \(H^\) (excitatory/inhibitory), into the gain factor \(M_k^\) for the k-th area as
$$\begin M_k^E = \left( 1 + b^E_ + s^E_ A\beta _k\right) \left( 1 + b^E_\tau + s^E_\tau tau_k\right) \end$$
(8)
$$\begin M_k^I = \left( 1 + b^I_ + s^I_ A\beta _k\right) \left( 1 + b^I_\tau + s^I_\tau tau_k\right) \end$$
(9)
where \(b^_\) are the excitatory/inhibitory A\(\beta\) and tau bias parameters, while \(s^_\) are the respective scaling factors. These are the 8 (from which actually only 6 are needed as tau only affects excitatory neurons [39], see next section) parameters that we will optimize for each subject individually.
ConstraintsBased on previous neuroscientific experiments [4], we included constraints on the direction of effect of A\(\beta\) and tau (i.e., inhibitory vs. excitatory influence). We introduced the following constraints:
A\(\beta\) produces inhibitory GABAergic interneuron dysfunction [6, 40], thus we can infer that \(s^I_ < 0\).
A\(\beta\) produces impaired glutamate reuptake [6, 40], so we can introduce the bound \(s^E_ > 0\).
Tau appears to target excitatory neurons [39], so we can safely consider that \(b^I_\tau = s^I_\tau = 0\).
Tau binds to synaptogyrin-3, reducing excitatory synaptic neurotransmitter release [41], thus \(s^E_\tau < 0\).
Although the interplay between A\(\beta\) and tau is not completely known [4], but there is evidence that A\(\beta\) promotes tau by cross-seeding [42, 43], thus the cross-term factors (i.e., the ones resulting from the multiplication of A\(\beta\) and tau scaling parameters) play a crucial role to elucidate the final impact of the combined burden.
ObservablesEdge-centric FCThe static edge-level FC is defined as the \(N\times N\) matrix of BOLD signal correlations between brain areas computed over the entire recording period (see Fig. 3). We computed the empirical FC for each human participant and for each simulated trial, as well as for the group-averages SC matrix of the healthy cohort. All empirical and simulated FC matrices were compared by computing the Pearson correlation between their upper triangular elements (given that the FC matrices are symmetric).
Fig. 3
Visualization of the SC graph, in matrix form (left) and as a graph showing the strongest 5% of connections. Node positions are computed with Fruchterman and Reingold’s [44] algorithm, which assumes stronger forces between tightly connected nodes. Besides the high degree of symmetry, we can observe the laterality is kept in the graph structure (also for subcortical regions). Node size linearly represents the graph theoretical measure of structural degree for each node. As we can see, the most important hubs are in the subcortical regions
swFCDThe most common and straightforward approach to investigate the temporal evolution of FC is the sliding-window FC dynamics (swFCD) [45]. This is achieved by calculating the correlation matrix, FC(t), restricted to a given time-window \((t-x:t+x)\), and successively shifting this window in time resulting in a time-varying \(FC_\) matrix (where N is the number of brain areas and T the number of time windows considered). Here, we computed the FC over a sliding window of 30 TRs (corresponding approximately to 1.5 min) with incremental shifts of 3 TRs. This FCD matrix is defined so that each entry, (\(FCD(t_x,t_y)\)) corresponds to the correlation between the FC centered at times \(t_x\) and the FC centered at \(t_y\). In order to compare quantitatively the spatio-temporal dynamical characteristics between empirical data and model simulations, we generate the distributions of the upper triangular elements of the FCD matrices over all participants as well as of the FCD matrices obtained from all simulated trials for a given parameter setting. The similarity between the empirical and model FCD distributions is then compared using the KS distance, \(D_\), allowing for a meaningful evaluation of model performance in predicting the changes observed in dynamic resting-state FC. However, the fundamental nature of the swFCD technique implies the choice of a fixed window len
Comments (0)