In 2014, a new Mendelian disease, SAVI, was classified as a novel type I interferonopathy, in which enhanced type I IFN (IFN-I) pathway activation plays a key role [1]. The IFN-I pathway produces innate immunity and antiviral proteins by sensing aberrant dsDNA via cGAS-cGAMP-STING axis, which activates IRF3 and IFN-I expression [4,5,6]. In addition, cGAS–STING also mediates NF-κB activation and interacts with various cell death and senescence pathways [4, 5].
Herein, we described a case of SAVI caused by the p.V155M heterozygous mutation of TMEM173, which was inherited from the female proband’s father. To our knowledge, more than 70 SAVI cases with autosomal-dominant SAVI have been published since 2014 [6]. Disease onset of SAVI usually manifests by the age of 1 year [7], ranging from the neonatal period to adulthood, though adult onset is rarely observed. Vasculopathy resulting from vasculitis and endothelial cell death is a hallmark of SAVI. The major clinical characteristics of SAVI are pulmonary involvement, skin disease, recurrent systemic inflammation, and developmental retardation. Other clinical features may overlap with monogenic interferonopathies to some degree [7]. Pulmonary involvement was noted in most studied patients (67–82%), which was the main reason for fatalities in SAVI [8, 9]. In our report, the proband and her father were found to have imaging changes in interstitial pneumonia. However, not all patients are symptomatic, as demonstrated by our case, wherein the proband experienced severe respiratory symptoms beginning at a young age while her father was not overtly symptomatic until an adult age. ILD was the prominent manifestation of pulmonary involvement. It occurred early in the disease and was initially insidious, characterized by early onset progressive dyspnea, tachypnea, and/or cough, resulting in pulmonary fibrosis and end-stage respiratory failure. Diffuse alveolar hemorrhage was also reported in the literature [8, 10], which was a main feature of COPA syndrome, another subtype of type I interferonopathy. Ground-glass opacities were the most typical lesions of chest CT, followed by crazy paving patterns, reticular opacities, and cysts. The lesions were frequently asymmetrical compared to other ILD lesions associated with CTD [6, 7, 11]. Pulmonary function tests usually show nonspecific effects, mainly exhibiting a restrictive pattern with diffusion impairment, but may also associate with an obstruction or a mixed lung function impairment [7, 9]. Pulmonary function tests were not performed on this patient due to her young age.
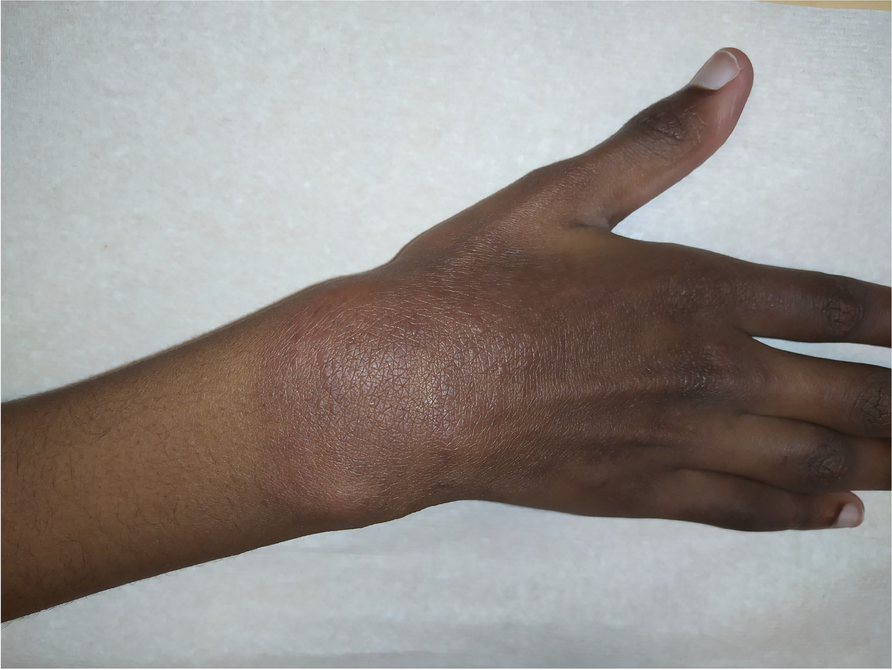
Skin disease of variable severity is displayed in 68.6–86% of patients [2, 7]. Severe skin vasculopathy can present in the nose, cheeks, ears, and acral zones, with extreme skin phenotypes involving perforation of the nasal septum, gangrene of the extremities, and extensive tissue loss. Mild lesions comprise acral and malar rash, purpuric rashes, chilblain lesions, telangiectasia, nail dystrophy, and livedo reticularis. Symptoms of alopecia, sparse and thin hair, and oral mucosal lesions were reported in some cases [7, 11]. Skin biopsies often reveal vasculopathy including perivascular inflammation and vasculitis [1, 12]. SAVI itself may carry an infectious susceptibility [12]. Failure to thrive is a common feature, as weight and height remained below standard deviations [7]. In our case, growth retardation was characterized by substandard height and weight, and skeletal dysplasia, such as delayed fontanel closure and pigeon breast deformity. Besides, articular manifestations are frequent in patients with positive RF. Bone destruction will develop as the disease progresses. In addition, other accompanying symptoms including recurrent febrile attacks, myositis, osteoarthropathy, neurological manifestations, hepatitis, cardiac disease (subepicardial ischemia, pericarditis), and kidney involvement (microscopic hematuria and mild proteinuria) were reported in sporadic cases [7,8,9, 11, 12].
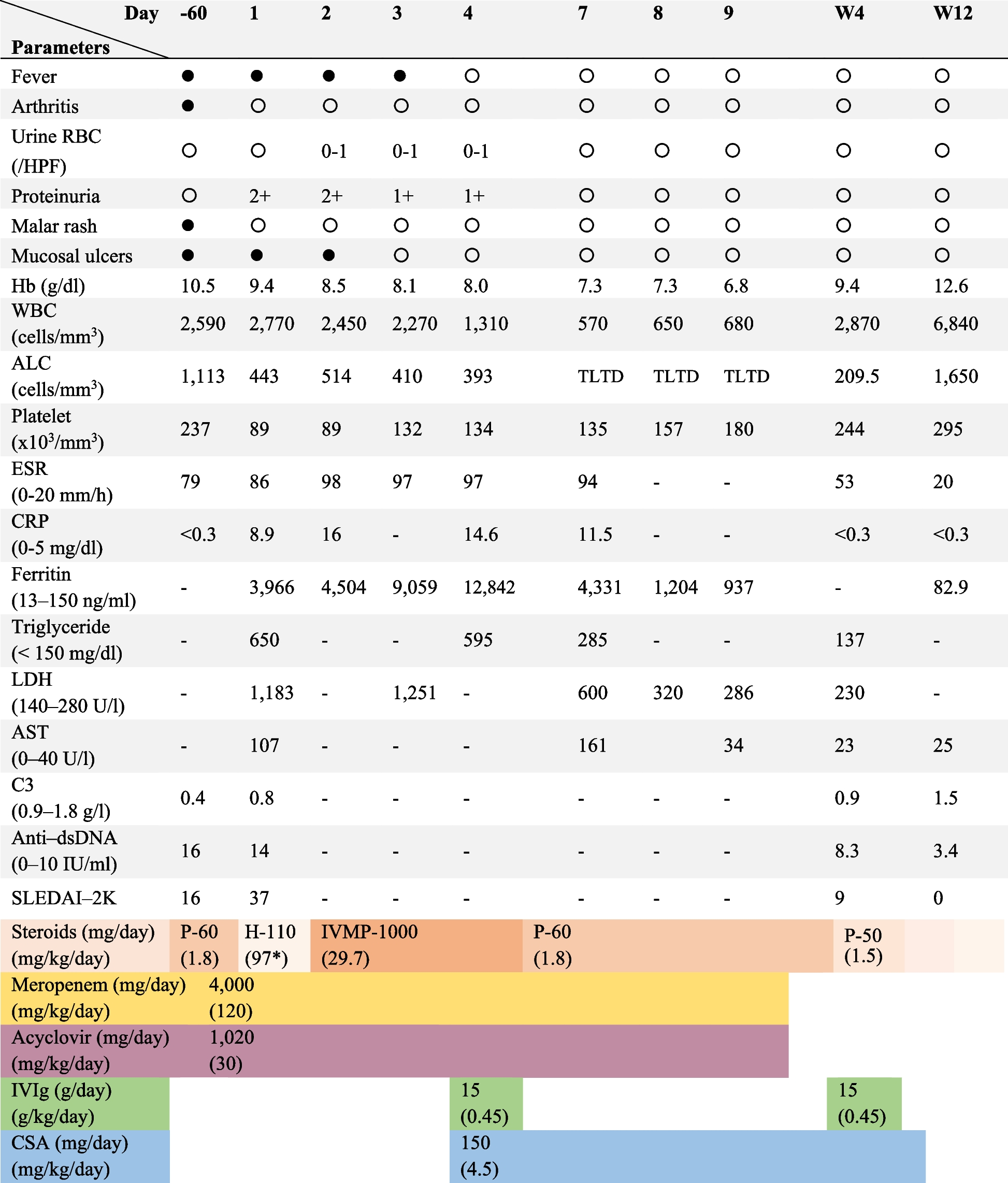
Laboratory tests of previous studies revealed that SAVI is characterized by systemic inflammation with increased levels of CRP and ESR, which may hardly return to normal [13]. Immune function tests demonstrated hyperimmunoglobulinemia, mainly hyper-IgG and IgA levels despite variable IgM levels. Complement levels were normal in most patients. Mild decreases in CD3 + and CD4 + T lymphocytes and elevations in CD19 + B lymphocytes were frequently observed, while decreased percentages of central memory CD8 + and effector memory CD8 + T lymphocytes with increased percentages of naïve CD4 + and CD8 + T lymphocytes were common [7, 8, 11]. Other common laboratory features of SAVI include positive autoantibodies, mostly high titers of ANA and ANCA with low specificity for MPO and PR3, followed by anti-cardiolipin antibody, lupus anticoagulant, anti-double-stranded DNA antibody, and anti-phospholipid antibody [12]. These autoantibodies highlight the early differential diagnosis with systemic lupus erythematosus or ANCA-vasculitis. Increased RF and CCP were documented, which, when complicated with arthralgia symptoms in early onset, may also confound the diagnosis [14]. High levels of IFN score and STAT1 phosphorylation were recorded in the tested patients [11]. The index case presented typical laboratory features and clinical symptoms. During the 14-month follow-up, the respiratory symptoms were improved, and the chest HRCT was not aggravated after the combined treatment of the baricitinib and steroids. However, improvement of laboratory indicators was limited, especially in autoimmune indices.
Since the first description of SAVI in 2014, a total of 19 types of pathogenic variants in STING1 including p.H72N, p.R94H, p.V147M/L, p.F153V/I, p.N154S, p.V155M, p.G158A, p. G166E, p.C206Y/G, p.G207E, p.R281W (homozygous mutation), p.R281Q, p. R284G/S, and p.K338Rfs*9 have been reported, with 2 (p.R94H, p.K338Rfs*9) of them expressed as uncertain significance [6, 15]. These mutations were heterozygous, and most of them were de novo. Previous reports suggested that SAVI remains to be reported as an exclusively autosomal-dominant disease [6]. However, recent reports have presented that autosomal-recessive inheritance caused by homozygous missense pathogenic variants (such as p.R281W) was found in STING1 [16]. Thus far, the genotype-phenotype correlations are not definite due to the limited cases. Jinying Li et al. [17] found that the phenotypes of SAVI in their study have considerable variability compared to the same mutation previously reported. Rohit G. Saldanha et al. [18] suggested that the severity and natural courses of the disease may not relate to mutation type. A recent literature review also returned no obvious phenotypic differences among mutations [7]. As for reasoning, Rensheng Wan et al. supposed that other factors (environmental factors, ethnic background, etc.) in addition to inheritance patterns that resulted in various genotypes-phenotypes should be considered [16]. Nevertheless, a few studies suggested that some clinical features of SAVI may be related to corresponding mutations. Several authors confirmed that patients with the inherited p.V155M variant have a less severe disease course than those with de novo variants [1, 16]. A systematic review demonstrated that, compared to patients with the p.V155M mutation, patients with p.N154S mutations had earlier disease onsets and more severe skin lesions, with no differences in respiratory symptoms [2]. These phenomena of heterogeneity on phenotypes were also observed in mouse models [19]. Our case demonstrates the phenotypic heterogeneity of the same genotype in pulmonary disease. The two patients have identical genotypes, but show distinct differences in the onset and progression of their pulmonary manifestations. The proband had early-onset and rapidly progressive pulmonary failure in childhood, while her father had late-onset and slowly progressive pulmonary lesions in adolescence. Moreover, the proband’s father did not suffer from any noticeable growth retardation, which differed from the proband’s phenotype. Above all, the relationships between genotype and phenotype remain for further study.
Currently, there is no standard treatment approach for SAVI. The treatment effects of immunosuppressive agents and biological therapies are mostly limited. Systemic corticosteroids are partially effective in SAVI, yet combination therapies with traditional disease-modifying anti-rheumatic drugs and IVIG had no significant additional effect [2, 11]. Besides, biological agents including TNF-α inhibitors, anti-CD20 antibody, anti-IL-6 inhibitor, and anti-BLyS antibody have disappointing efficacy in SAVI [12, 14]. Based on the disease pathogenesis and the understanding that IFN-I signaling upregulation subsequently activates the JAK /signal transducer and activator of transcription (STAT) pathway, JAK inhibitors have been raised as new potential treatment options. The first use of a JAK inhibitor (ruxolitinib, a selective JAK1/2 inhibitor) in SAVI was reported in 2016 in three cases, which resulted in marked positive effects on skin symptoms, CRP level, interstitial pneumonia, and pulmonary function [20]. Subsequently, the treatment of SAVI with JAK inhibitors has been brought to the forefront and evaluated in more case reports. An overviewing report found that SAVI patients undergoing ruxolitinib treatment before the occurrence of irreversible damage improves clinical features of the disease, especially lung damage [7, 21]. However, there is no consensus on the efficacy of ruxolitinib. Combined treatment of corticosteroids with ruxolitinib in a case report demonstrated rapid improvements in pulmonary hypertension and general well-being as well as resolution of the IFN gene signature, but such treatment coincided with the progression of nasal septal erosion [18]. Moreover, Yan Wang et al. [11] reported that two patients who underwent ruxolitinib treatment exhibited poor responses including a fatality. They speculated that the moribund patient may have succumbed due to the severe disease status and the late introduction of ruxolitinib. Another case with sustained elevation of circulating inflammatory cytokines may have resulted from insufficient dosage. Another selective JAK1/2 inhibitor, baricitinib, was also assessed in SAVI patients. In four cases, baricitinib treatment improved the skin flare, inhibited the further loss of digits, stabilized the ILD and respiratory function, and reduced the requirement of glucocorticoids, while inflammatory markers did not decrease to normal [3]. A recent case report found that baricitinib treatment may have a mitigating effect on disease phenotype, but it failed to prevent further progression of the disease and improve the patients’ IFN signature [16]. Of note, the half-life of baricitinib, the dosage, and the time of administration should be considered when evaluating treatment effects [22, 23]. Baricitinib was chosen in our case and showed some effect on ILD and inflammatory index. However, compared to JAK1/2 inhibitors, the JAK1/3 inhibitor tofacitinib seems to be less effective. Wendao Li et al. [13] reported two SAVI cases treated with baricitinib and tofacitinib, respectively. They both showed an improvement in the disease scores, but baricitinib treatment resulted in a dramatic improvement in lesions of diffuse cords, patchy consolidation, and ground-glass opacities while tofacitinib treatment did not [13]. Although a marked suppression of the IFN signature and therapeutic effects on skin lesions were observed after treatment with tofacitinib [24], the pulmonary defects remained unchanged [25]. Moreover, Xiaolei Tang et al. described two SAVI patients with p.V155M mutations receiving combined treatment of tofacitinib with unsatisfactory consequences [8]. Poor therapeutic effects were observed in improving rashes and ILD and reducing the dosage of steroids. Another patient with early onset SAVI who received short-term tofacitinib treatment died at the age of five months due to respiratory failure [26]. An in vitro study confirmed that tofacitinib could not inhibit dsDNA-triggered, STING-dependent IRF3 phosphorylation in a SAVI model, which suggested that it may not provide an optimal therapeutic intervention to prevent STING-related disease [27]. These studies provide various responses of SAVI patients to JAK inhibitors. The other hypothesis regarding a linkage between the treatment response and a given mutation type needs to be queried in the future, as more SAVI cases will undoubtedly be reported. In addition, we should pay attention to the higher risk of viral infections under the use of JAK inhibitors, as it might aggravate pulmonary fibrosis [16, 28]. Besides, other therapeutic strategies including monoclonal antibodies to IFN-I receptors and STING inhibitors might provide promising therapeutic perspectives [6].
In summary, we describe a case diagnosed as SAVI with severe ILD, which remains stable under the combination therapy of baricitinib and steroids. It seems that baricitinib expressed a therapeutic effect on SAVI in some ways. However, the lung CT image shows no obvious improvement. In general, based on previous literature, JAK inhibitors failed to control the inflammation of the disease completely, which calls for further exploration of JAK inhibitors or other therapeutic strategies, such as monoclonal antibodies to IFN-I receptors or STING inhibitors, to more optimally treat this inflammatory disease.



Comments (0)