
Remember me


Molecular and bioinformatic-based studies are often focused on identifying the presence of dysregulated biological processes and pathways in rheumatic disease. Sparks et al. reframed this question and instead asked what constitutes immune health [1]. Through integrating and analyzing whole blood transcriptome, serum proteome and immune cell frequency data across a cohort of 228 patients with various monogenic immune diseases and 42 healthy controls, a quantitative immune health metric (IHM) was developed. The IHM is composed of multiple features, including more traditional inflammatory and myeloid cell signaling proteins (MIP1a, IL18R1) as well as NK cell frequency, but also lesser considered contributors to immune health, such as transcription factors like eIF4A3. The IHM can potentially be used as a biomarker of disease activity and treatment response across multiple diseases. As related to the field of Pediatric Rheumatology, the IHM was discovered to be lower with higher disease activity in a cohort of pediatric systemic lupus erythematosus patients.
A (mouse) cytokine response dictionaryCytokines don’t exist just to confuse medical students [2], they orchestrate and regulate immune responses in health and disease. Adding to an increasingly rich and important body of “omic” resources [3], Cui et al. performed single-cell RNAseq on lymph node cells following systemic administration of 86 cytokine combinations [4]. They curated responses of 17 immune cell types, and found that certain cytokines (IL-2, IL-12, IL-18, IL-15, IFNg) resulted in an overall similar pattern of responses. IL-1β uniquely executed a different transcriptional pattern in nearly every cell type examined. Rare immune cell types often demonstrated the most robust increases in cytokine gene expression. They built a computational tool, the Immune Response Enrichment Analysis (IREA), that can be used to assess for cytokine signatures or immune cell polarization in other investigators’ datasets.
Our massive immune systemsWhile all cells are immune cells, we tend to favor cells of hematopoietic (and sometimes yolk sac) origin as more relevant for the study of immune dysregulation. Sender et al. performed an estimation of the number and mass of immune cells in the typical adult human, with some unexpected insights [5]. Though the gastro-intestinal tract is often considered the largest “immune” organ, of our estimated 1.8 × 1012 immune cells, 39% reside in lymphatic organs (including the spleen), and both gastrointestinal (GI) tract and lymphatic organs were dominated by T, B, and plasma cells. Another 39% reside in the bone marrow, dominated by a tremendous reservoir of neutrophils. Mast cells, often overlooked as esoteric “allergy” cells not present in blood, constituted up to 30% of the “immune” cells of the skin or lungs.
Neuroimmunity (because immunology was getting too simple)It is increasingly obvious that the central and peripheral nervous systems contribute to adaptive and innate immune host defense. Three manuscripts beautifully illustrate this concept and how understanding the specifics may foster new diagnostic and therapeutic strategies.
In trying to understand how the brain contributes to controlling systemic inflammation, Jin et al.. demonstrated how both pro- and anti-inflammatory cytokines, via different types of vagal nerves (CALCA and TRPA1, respectively), acted via the central nervous system to coordinate an appropriately calm peripheral inflammatory response [6].
Patients with pain sensitization disorders are often among the most disabled by their disorder to appear before a Pediatric Rheumatologist, making the complex interplay between inflammation and pain sensitization a pressing problem. Jain et al. found that macrophage responses to inflammatory signals could either promote (via prostaglandins) or inhibit (via thrombospondin) pain-sensing nerve hypersensitivity, providing a possible means of preventing the development of pathologic pain syndromes [7].
The gut is a large immune organ, and critically regulated by the enteric nervous system, with implications particularly for inflammatory bowel disease (IBD). Zhu et al. used a chemogenetic screen to show that signals from afferent Trpv1-expressing nociceptive nerves, acting through dorsal root (but not vagal) ganglia, inhibited a critical population of gut-resident CD4 + regulatory T-cells (Treg) [8]. Inhibition of this pathway may stabilize gut Treg and offer a novel IBD treatment approach.
Promising new technologiesAmongst a plethora of advances, a few novel technologies promise to change how we study, understand, and possibly treat PedRheum diseases.
Given the outsize importance of monogenic variants in PedRheum diseases, Schmidt et al..’s demonstration of base-editing (targeted changes in DNA sequence) in primary human T-cells may revolutionize how we study the effects of specific genetic variants [9].
For years investigators have had the ability to identify clonal T-cell populations in cancer. In autoimmunity, clonal proliferations of CD4 T-cells could be autoreactive or regulatory, but their peptide specificity remained mysterious. Dezfulian et al. created a platform called TScan-II that enabled them to identify the peptides associated with CD4 T-cell clones. This enables investigators to ask a host of new questions relevant to the nature of maintaining and breaking tolerance.
PedRheum owes a great debt to insights gleaned from animal models, but understanding these has been hampered by the challenge of tracking cell-cell interactions in vivo. Nakandakari-Higa et al. developed a universal version of their Labeling Immune Partnerships by SorTagging Intercellular Contacts (uLIPSTIC) to enable the temporal tracking of cells with whom your cell-of-interest has significantly interacted [10].
Combining single-cell sequencing with sequential intravascular labeling of immune cells in a glioblastoma mouse model, Kirschenbaum et al.., have taken another step in integrating the variable of time to single-cell systems-immunology approaches to immune responses. [11].
Spatial transcriptomics technology has also been rapidly evolving even over the last year. The ability to better understand cell-cell interactions and in particular tissue-resident-immune cell interactions at single-cell spatial resolution has the potential to pave the way toward precision medicine in PedRheum diseases. As an example toward insights spatial transcriptomics technology can provide within PedRheum diseases, Danaher et al.. utilized the CosMx platform to evaluate diagnostic kidney biopsies from 7 children with proliferative lupus nephritis (LN) and 4 non-inflammatory controls, identifying increased myeloid cell populations within LN glomeruli and variable macrophage gene expression based on spatial localization, perhaps suggesting emerging connections of macrophage tissue localization to function and disease pathogenesis [12]. Additional investigation into three serial kidney biopsies from a single treatment refractory pediatric LN patient revealed persistent tubulointerstitial B-cells despite aggressive immunosuppression with the B-cell depleting agent rituximab as well as cyclophosphamide and multiple other agents, highlighting the importance of understanding how to target tissue-based as well as circulating cells.
New mechanistic insights into pedrheum diseases Linking infectious response to development of multisystem inflammatory syndrome in children (MIS-C)A subset of children infected with SARS-CoV-2 develop a severe, post-infectious complication termed MIS-C; yet, the mechanistic link leading to MIS-C development in only some children is unclear. Bodansky et al. utilized a technique called phage immunoprecipitation sequencing (PhIP-seq) to identify autoreactivity of patient antibodies to the human and SARS-CoV-2 proteomes [13]. Notably, children previously infected with SARS-CoV-2 with development of MIS-C had enrichment for 107 autoantigens, including sorting nexin 8 (SNX8), as well as a specific epitope within the SARS-CoV-2 proteome, termed the MIS-C associated domain of SARS-CoV-2 (MADS). The immunoreactive regions of SNX8 and MADS were demonstrated to have high sequence similarity, and children with development of MIS-C also displayed cross-reactive CD8 + T-cells specific for SNX8. These identified T-cells were further able to cross-react with the SARS-CoV-2 MADS region, suggesting that cross-reactive CD8 + T-cells may drive MIS-C pathogenesis in a subset of children after SARS-CoV-2 infection.
Was there MIS-C before COVID? Interestingly, syndromes with a clinical phenotype similar to MIS-C have previously been described in children, such as Kawasaki’s disease shock syndrome. Benezech et al. reported expansion of Vβ21.3 + T-cells in two separate cohorts of children pre-COVID-19 pandemic, including (1) children admitted to the ICU with a clinical phenotype resembling MIS-C, with later viral testing indicating positive antibodies for coronaviruses, including the seasonal human coronavirus 229E in one patient, and (2) children with a diagnosis of Kawasaki’s disease shock syndrome [14]. These findings indicate that MIS-C may be a broader pediatric syndrome that can be triggered by multiple infectious agents.
Activated CD8 T-cells as biomarkers and pathogenic in MAS and HLHHemophagocytic Lymphohistiocytosis (HLH) refers to a physiotemporal cytokine storm syndrome of fever, cytopenias, coagulopathy, hepatosplenomegaly, hepatitis, CNS inflammation, and potential for multi-organ dysfunction and death. Macrophage Activation Syndrome (MAS) refers to this syndrome occurring in the context of a rheumatic disease, most specifically Still’s Disease (a.k.a. either Systemic JIA (sJIA) or Adult-onset Still’s disease (AOSD)) [15], but HLH occurs in infectious, malignant, iatrogenic, and monogenic contexts as well. These monogenic forms of HLH, specifically defects in Perforin or other proteins necessary for granule-mediated cytotoxicity, have long identified Cytotoxic T Lymphocyte (CTL) dysfunction as a potential driver of familial HLH. In this Perspectives series, Lam et al. [16] more closely overviews the many recent studies that implicate highly activated T-cells, specifically CD38+HLA-DR+CD8 T-cells, as a potential biomarker and unifying pathogenic mechanism in familial HLH [17, 18], MAS [19, 20], and HLH-like presentations of liver failure [21], [22], [23]. Such cells may also be part of typical immune responses to certain HLH-prone infections, like EBV [24, 25].
Mitochondrial nucleic acids not only drive interferon response, but also IL-1β secretion in systemic lupus erythematosus (SLE)The role of mitochondrial nucleic acids (mtNAs) in contributing to autoimmune disease pathogenesis is increasingly being recognized, but disease-specific mechanisms and relevant associated signaling pathways need to be further delineated [26, 27]. Caielli et al. noted that active SLE patients with presence of red blood cells with retained mitochondria (Mito + RBCs) also have an expansion of IL1β + MxA + monocytes [28]. To better understand the mechanism leading to interferon (IFN)-stimulated gene and IL1β expression on monocytes, Mito + RBCs were generated that were deficient in mitochondrial nucleic acids. Monocyte stimulation with Mito + RBCs deficient in mtNAs resulted in decreased IFN-stimulated protein and IL1β production, highlighting the critical role of mitochondrial nucleic acids for both IFN response and IL-1β production from monocytes. Monocyte-derived mtDNA was then identified as a trigger for IL-1β production, and MxA enabled IL1β translocation and secretion.
Calprotectin-mediated thrombocytopenia in antiphospholipid syndrome (APS)Pediatric APS often manifests with thrombocytopenia as an extra-criteria manifestation and can associate with higher risk of future thrombosis. Sloan et al. identified calprotectin, a heterodimer of S100A8 and S100A9, to be elevated in primary pediatric APS patients and to also negatively associate with platelet count [29]. Treatment of platelets with plasma from calprotectin-high vs. calprotectin-low APS patients was additionally found to decrease platelet viability. Hoy et al. further identified that there was a synergistic effect on platelet viability as well as platelet caspase-1 activity after concurrent treatment with both calprotectin and APS IgG [30]. Additionally, inhibition of TLR4 with Paquinimod and NLRP3-mediated inflammasome activity with MCC-950 rescued platelet viability. In conclusion, circulating calprotectin levels may indicate risk for thrombocytopenia and potentially subsequent risk for thrombosis in APS patients.
UNC93B1 links TLR7 and TLR8 with Monogenic lupusOne of the unique contributions of the pediatric rheumatologist is our experience with so-called Inborn Errors of Immunity (IEI), which provide concrete links between specific genes and complex inflammatory phenotypes. Perhaps no phenotype is more complex than that of SLE, and the tableau of genes with high-penetrance associations with SLE includes classical complement components (e.g. C1Q), DNAses (e.g. TREX1 & DNASE1L3), signaling molecules (e.g. TNFAIP3, PTPN2), and the endosomal nucleic acid sensor TLR7. While TLR7 gain of function is known to cause a SLE-like IEI [31], the specificity for this link– especially relative to other endosomal PRRs like TLR9– remains unknown.
Over the past year, several groups identified variants in UNC93B1 leading to hyperactivity of TLR7 and/or TLR8 (Table 1) [32, 33, 34, 35, 36]. Though phenotypes were distinctly SLE-like, variants that led to hyperactivity of only TLR8 were conspicuously associated with Chilblain’s-like cutaneous features, but not the systemic autoimmunity associated with variants that also affected TLR7 activation. UNC93B1 is a chaperone and scaffolding protein that assists in both TLR transport and proper TLR configuration in the endosome. Wolf et al. determined that a specific UNC93B1 variant associated with early-onset SLE led to decreased stability of the UNC93B1 protein and subsequent destabilized interaction with TLR7, resulting in TLR7 hyperactivation [32]. Additional UNC93B1 variants leading to TLR7 hyperactivation by other mechanisms were soon discovered, including an UNC93B1 variant that results in endosomal TLR7 build-up as a result of reduced interaction with BORC (biogenesis of lysosomal organelles complex-1 related complex) [33]. BORC is needed for TLR7 receptor turnover and degradation; therefore, reduced interaction of TLR7 with BORC or BORC deficiency leads to accumulation of TLR7.
Table 1 UNC93B1 variants leading to hyperactivity of TLR7 and/or TLR8 PTPN2 haploinsufficiency also leads to development of SLE and Evan’s syndromePreviously associated with autoimmune enteropathy, combined immunodeficiency and multi-organ autoimmunity [37, 38], JeanPierre et al. described six novel variants in protein tyrosine phosphatase non-receptor type 2 (PTPN2) in children with lupus and Evan’s syndrome [39]. PTPN2 plays a role as a negative regulator of JAK-STAT signaling, and loss of PTPN2 protein expression leads to JAK/STAT pathway hyperactivation and presentation of clinical autoimmune disease features. This highlights that it is equally as important to consider negative regulators of cytokine and immune pathways and their role in preventing hyperactivation in the development of autoimmune disease. We can also consider mimetics of negative regulators as possible alternate treatments to develop in PedRheum patients.
Ubiquitinopathies and autoinflammatory diseaseUbiquitylation is a necessary post-translational modification to regulate protein stability, function, signaling and degradation. As ubiquitylation is also essential for innate immune response regulation, there have been autoinflammatory disorders described as ubiquitinopathies. Davidson et al. recently described a dominant negative mutation in OTULIN, a deubiquitinating enzyme, that led to OTULIN-related autoinflammatory syndrome (ORAS), manifested by fever, cutaneous inflammation and lung involvement in the setting of an elevated type I IFN signature and increased sensitivity to TNF-induced cell death [40]. A loss of function mutation in SHARPIN, a component of the linear ubiquitin assembly complex, was also identified as a new ubiquitinopathy, with clinical manifestations of recurrent fever, parotitis, arthritis and colitis [41].
Novel therapeutic targets and treatment approaches in rheumatic disease Non-inflammatory pathways for therapeutic targeting in myositisNon-immune mechanisms may contribute more to disease chronicity and immune activation in PedRheum than we currently account for. Abad et al. sought to evaluate mechanisms contributing to development of myositis in an inducible T cell co-stimulator (Icos) knock out (KO) Non-Obese Diabetic (NOD) mouse model. Myositis develops at about 25 weeks of age in this model, with muscle tissue displaying elevation of both IFNβ and IFNγ gene expression [42]. Muscle proteomic analysis in these mice revealed a theme of metabolic signaling pathway downregulation, and the majority of dysregulated proteins localized to mitochondria. Icos KO NOD mice with myositis also displayed increased expression of oxidative response genes in muscle, and muscle tissue homogenates produced more reactive oxygen species as compared to Icos+/+ NOD mice. Treatment of mice with both N-acetyl cysteine (NAC) and anti-IFNγ treatment led to improved strength and decreased inflammatory infiltrate in muscle tissue. Based on this study and also other work describing oxidative stress and mitochondrial dysfunction in juvenile dermatomyositis [43, 44], it is possible that targeting reactive oxygen species or mitochondrial dysfunction may have a role in treatment of myositis.
Glucocorticoids and macrophage mitochondrial metabolismDespite the essential role played by glucocorticoids (GC) in the current management of inflammatory and immune-mediated disease, their mechanisms of action remain mysterious. Auger et al. identified an acute effect of glucocorticoids on the induction of itaconate, a TCA-cycle intermediate with potent anti-inflammatory effects in myeloid cells like macrophages [45]. This effect required the glucocorticoid receptor (GC-R), but not new transcription. Instead, the activated GC-R interacted with the mitochondrial pyruvate dehydrogenase complex, driving TCA flux and aconitate decarboxylase 1 (ACOD)-dependent itaconate production. This mechanism was crucial for GC’s ability to restrain models of innate, autoimmune, and allergic inflammation.
Deeper B-cell depletion with immune effector cells (IECs)For the first time in decades, clinicians and investigators are beginning to entertain the notion of a cure for autoimmunity. The idea that B-cell depletion might treat autoimmune disease is as old as their connection to autoantibodies. Borrowing tools from oncology, the first experience with rituximab in Rheumatoid Arthritis is now more 20 years old [46]. The use of B-cell depleting antibodies has become widespread in autoimmune diseases, but despite rapid and robust elimination of peripheral B-cells, durable remissions have been rare. Borrowing again from oncology, the avalanche of case series reporting almost “too-good-to-be-true” efficacy of T-cell dependent B-cell depletion for patients with refractory autoimmunity may represent an inflection point in the history of our field. The clinical details of these reports are beyond the scope of this Perspective. They derive mostly from Chimeric Antigen Receptor T-cells (CART) directed against B-cell antigens like CD19 and BCMA in treatment refractory SLE, inflammatory myositis, and scleroderma [47]. In PedRheum, case reports in refractory Juvenile Dermatomyositis [48] and SLE nephritis [49] have been similarly positive. Autologous CART are difficult and costly to make, and patients must receive chemotherapeutic lymphodepletion to “make room” for the CART. It is therefore reassuring that early experience with engineered proteins that biochemically link B-cell antigens to T-cell signaling molecules termed bispecific T-cell engagers or BiTEs (namely CD3ζ, e.g. blinatumomab and teclistamab) have been similarly impressive [50, 51, 52].
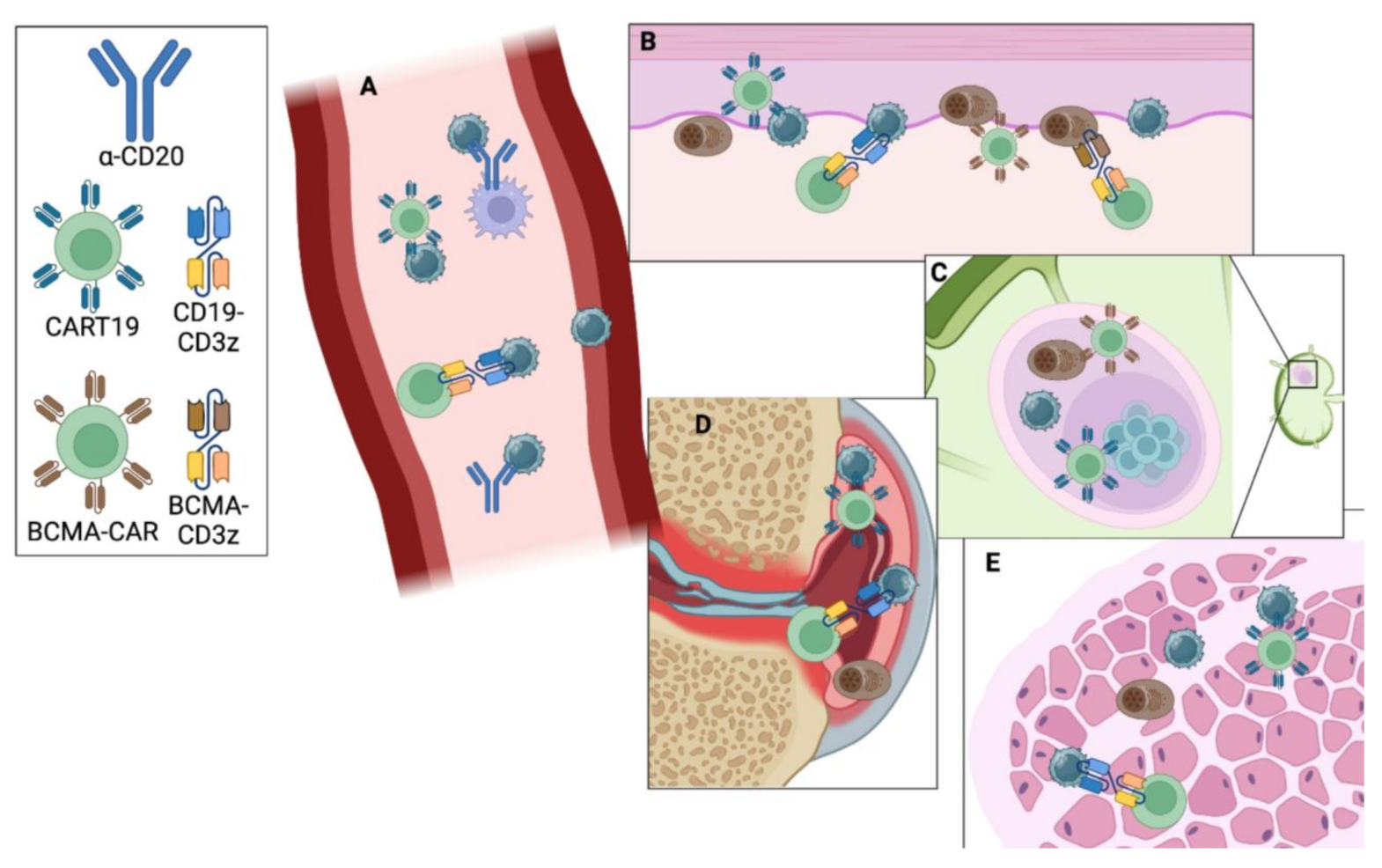
IEC therapies result in rapid peripheral B-cell depletion, and like B-cell depleting antibodies, the B-cell compartment begins to repopulate about 3 months after IEC treatment. By contrast, the B cells that begin to emerge following IEC treatment are overwhelmingly naïve. When viewed alongside profound & durable clinical improvement and autoantibody normalization, this suggests that deeper B-cell depletion may provide the autoreactive B-cell “reset” that rheumatologists having been seeking for decades. The mechanism for this deeper depletion likely rests with T-cells’ expertise in infiltrating tissues, expanding in response to TCR stimulation, and killing multiple targets. That IEC therapies target B-cell antigens expressed throughout the B-cell life cycle and/or in plasma cells (e.g. CD19 & BCMA, respectively) that may also contribute to their greater depth of depletion. It remains an open question whether IEC therapy is also a “reset” in terms of B-cell memory to pathogens, in part due to the widespread availability of immunoglobulin replacement. In short, it appears that B-cells are the “bad guys” in autoimmunity, but effectively depleting them requires recruiting expert killers (Fig. 1).
Fig. 1
Antibody versus Immune Effector Cell (IEC) Strategies for B-cell Depletion: Antibodies like rituximab and obinutuzumab bind CD20 on B-cells and target them for death via phagocytosis by macrophages or antibody-dependent cellular cytotoxicity by NK cells. This is efficient in peripheral blood (A). By contrast, CART and bi-specific antibody strategies rely on T-cells and appear to be effective at depleting B- and plasma cells in blood as well as skin (B), lymph node (C), synovium (D), muscle (E), etc
Comments (0)