3.1 MAA Evidence and Results
For 36 mAbs and fusion protein biosimilar candidates (mostly IgG1), the quality/CMC (i.e. general quality aspects and analytical comparability exercise), clinical PK/PD and clinical efficacy, safety and immunogenicity (E/S/I) aspects were analysed based on the information provided in the EPAR. Results are shown in Fig. 1 according to five possible scenarios.
For more than 80% of the biosimilar candidates analysed (29/36), the quality/CMC part of the dossier, the clinical PK/PD as well as the E/S/I results all unambiguously supported biosimilarity (Fig. 1, Scenario 1). For two biosimilar candidates, differences in some QAs and functional assays were observed [14, 15], but these differences were not seen in PK/PD and clinical E/S/I studies. One candidate had higher immunogenicity [15], later deemed irrelevant (see Discussion). All these biosimilar candidates listed for Scenario 1 obtained a MA.
Scenario 2 applies to two cases with an unsatisfactory quality/CMC package but with overall acceptable clinical trial results (Fig. 1, Scenario 2). In these two cases [43, 44], major concerns were raised regarding the biosimilarity exercise as well as regarding the comparability of the clinical batches and the commercial batches of the biosimilar. The clinical PK and efficacy trials formally met their primary endpoints. However, uncertainties remained for the clinical efficacy trial regarding secondary and subgroup analyses for the rituximab biosimilar candidate [43]. Both applications were withdrawn by the companies owing to major remaining uncertainties expressed in unresolved quality MO.
Scenario 3 was defined as those product candidates having an acceptable quality/CMC package but indicating differences in the clinical PK/PD profile or remaining questions regarding representativeness of test material used in the PK study, while all other clinical data demonstrated comparability (Fig. 1, Scenario 3). Two of the biosimilar candidates analysed [45, 46] had an initially failed PK study. In both instances, it was argued that the observed differences in glycan structures known to affect PK (high mannose content) were too small to explain the initially observed PK differences [58]. The conduct of a second PK trial with improved design features was requested and led to successful demonstration of similar PK profiles [59]. For a third biosimilar candidate, PK results were not accepted because the test product was not deemed representative of the commercial product [47].
Scenario 4 lists those cases with an acceptable quality/CMC package and successful PK trial but with issues regarding the clinical E/S/I package (Fig. 1, Scenario 4). For both affected trastuzumabs [48, 49] the primary efficacy endpoint was formally not met as the upper limit of the confidence interval (CI) was not contained within the pre-defined equivalence margin. For both trastuzumabs, a MA was granted based on the convincing quality/CMC, PK, safety and immunogenicity data packages, despite a failed primary endpoint.
The last hypothetical scenario would be unconvincing quality/CMC data and failed clinical trials (PK and efficacy trial), which was not observed in any of the 36 cases (Fig. 1, Scenario 5).
3.2 Analysis of First Regulatory Assessment Reports
For the majority of biosimilar candidates analysed (34/36), the LoQ raised by the CHMP in the D120 AR was adequately addressed by the applicants and thus led to the final approval.
Analysing the number of MO for the 36 biosimilar candidates concerning scientific issues indicates that 56% of MO were related to quality/CMC, 19% to clinical PK/PD and 25% to clinical E/S/I issues, respectively (Fig. 2a). Within the quality/CMC part, the majority of MO dealt with general pharmaceutical issues rather than biosimilarity aspects (Fig. 2b).
Analysis of OC revealed a similar distribution with 64% of OC pertaining to the quality of the biosimilar candidates, 12% to PK/PD and 24% to E/S/I (data not shown).
When categorising the 36 biosimilar candidates based on where MO were raised, i.e. quality versus PK/PD versus E/S/I, we differentiated four cases, depending on whether MO were identified and knowing that any unresolved MO would prevent approval. We differentiated case 1 when assessment of quality and clinical parts of the dossier led to no MO (positive alignment) in 42% (15/36) of the MAAs analysed, thus supporting biosimilarity. Case 2 was when the quality assessment led to MO that, if not resolved, would lead to rejection of the filing. This applies to 11% (4/36) of MAAs analysed. For case 3, when quality assessment supported biosimilarity but clinical queries challenged the validity of the package, 22% (8/36) of cases were identified (8% of cases with MO regarding PK/PD, 11% with MO regarding E/S/I and 3% regarding both PK/PD and E/S/I). And finally, case 4, when both quality and clinical packages raised concerns (negative alignment), with 25% (9/36) of MAAs analysed (Fig. 2c).
The main reasons for MO are summarised in Table 1.
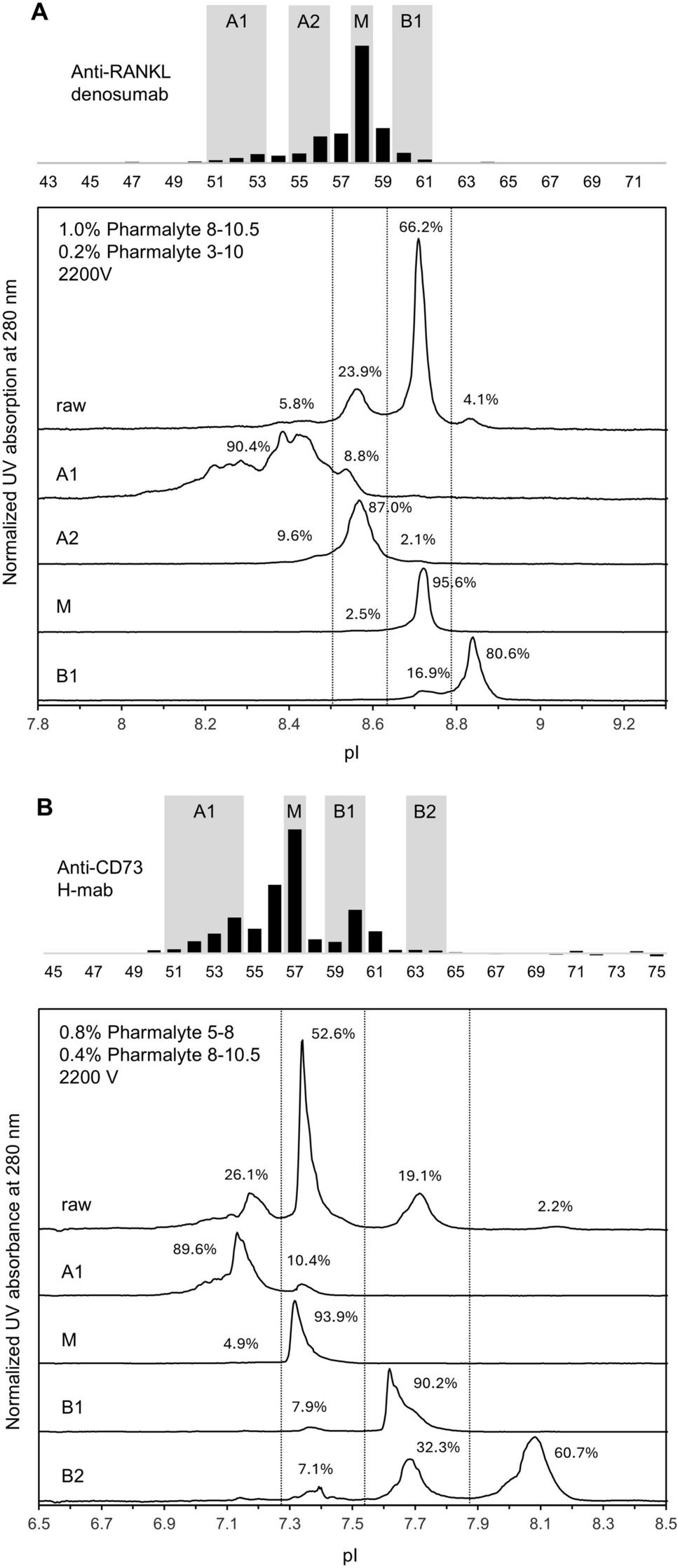
3.3 Evaluation of Analytical Biosimilarity and Clinical Comparability for Rituximab and Trastuzumab Biosimilars
Rituximab and trastuzumab biosimilar products were selected for further in-depth analysis of quality/CMC (Online Resource 1, Table 2; Fig. 3) and clinical data (Online Resource 2–5, Table 3) as these included withdrawn applications.
3.3.1 Comparison of Analytical Biosimilarity Across Products
The number of biosimilar batches analysed per product varied between 3 and 40, for most QAs. The analytical comparability packages of the rituximab and trastuzumab biosimilars comprised between 35 and 85 individual assays per product. For most of the QAs, orthogonal analytical methods were used.
Rituximab is an IgG1 kappa type mAb directed against CD20 expressed on the surface of pre-B and mature B lymphocytes, but not on hematopoietic stem cells and terminally differentiated antibody-producing plasma cells or other tissues. Upon binding to CD20, rituximab mediates B cell lysis (leading to B cell depletion) by three distinct mechanisms of action (MoAs): complement dependent cytotoxicity (CDC), antibody dependent cellular cytotoxicity (ADCC) and apoptosis [60]. Therefore, the biological activity of rituximab is determined by a combination of CD20 binding assay and an apoptosis induction assay, together with fragment crystallisable (Fc) functionality. Besides activating the pathways of CDC and ADCC, binding of rituximab to its target (CD20 expressed on B cells) also triggers apoptosis via the caspase signalling pathway [61]. Antibody-dependent cellular phagocytosis (ADCP) has been further implicated as plausible MoA of rituximab in its killing of chronic lymphocytic leukaemia cells [60, 62].
Trastuzumab is an IgG1 mAb which binds to human epidermal growth factor receptor 2 (HER2), a transmembrane oncoprotein overexpressed in approximately 20–25% of invasive breast cancers [63]. Binding of trastuzumab to HER2 inhibits ligand-independent HER2 signalling and prevents the proteolytic cleavage of its extracellular domain, an activation mechanism of HER2. As a result, trastuzumab inhibits the proliferation of human tumour cells that overexpress HER2. Therefore, the biological activity of trastuzumab is determined by the combination of HER2 binding assay and an inhibition of cellular proliferation assay, together with Fc functionality. However, in contrast to rituximab, CDC activation is not thought of as a MoA of trastuzumab [64].
For other fragment antigen binding (Fab) mediated assays, glycan and purity profile and charge variants we followed a similar categorisation as in our previous paper [13]. Additional assays include, for example, ADCP for both rituximab and trastuzumab, and inhibition of vascular endothelial growth factor (VEGF) secretion for trastuzumab.
Online Resource 1 provides a summary of the analytical biosimilarity results for approved rituximab (products A–C) and trastuzumab (D–I) biosimilars, and Table 2 provides a summary of the instances where less than 100% of batches were within the reference range and how the resulting uncertainty was resolved.
High similarity [≥ 90% of batches within range (solid dark and light-green horizontal stripes)] was found for protein content, biological activity (CD20 binding and apoptosis induction for rituximab, and HER2 binding and inhibition of cellular proliferation assay for trastuzumab), FcγRIIIa binding, neonatal Fc Receptor (FcRn) and C1q binding, ADCC and CDC for almost all rituximab and trastuzumab biosimilars. Exceptions included inhibition of cellular proliferation for one trastuzumab (product I), FcRn for one rituximab (product C) and one trastuzumab (product I) biosimilar and the high affinity FcγRIIIa v/v genotype for one rituximab (product B). However, as seen in Table 2, in most cases these differences were considered within the method variability or viewed as sufficiently justified based on high similarity found in other critical QAs (CQA) (i.e., ADCC for FcγRIIIa v/v), the results from PK comparability studies and regulatory experience. None of the authorised trastuzumab biosimilars displayed CDC activity (represented as dark-green vertical stripes in Online Resource 1), which is expected.
More variability was found for binding to other Fcγ receptors, purity and glycosylation profile, charged variants and additional assays (Online Resource 1). Again, as seen in Table 2 the observed differences in Fc binding assays and the glycan profiles were accepted because similarity was confirmed in biological assays. Moreover, afucosylation was 100% within range for all except one trastuzumab biosimilar (product G). Differences in purity and charge variants were seen as negligible based on regulatory experience and product understanding, and differences in additional assays were accepted based on the totality of the evidence presented for similarity.
3.3.2 Comparison of Analytical Biosimilarity for Withdrawn Products
Figure 3 represents the key quality/CMC requirements and whether these were met for the two withdrawn biosimilar applications [43, 44]. These key requirements were categorised following the classification from Bielsky et al. [56].
Of the quality/CMC requirements included, less than half were met for either of the products. Regarding the RP characterisation, both applicants failed to demonstrate two out of four of the prerequisites, demonstrating in both cases an in-depth knowledge of MoA and CQA of the RP but failing to analyse enough representative RP batches or to adequately establish the quality target product profile (QTTP). Regarding the biosimilar candidate attributes, out of the three prerequisites, only one was met for each product. The quality/CMC package included suitable and qualified analytical methods for the withdrawn rituximab and an adequate manufacturing process for the trastuzumab. However, none of the other requirements were met, including the representativeness of clinical and commercial batches or the use of additional orthogonal assays. Finally, only the withdrawn rituximab included an adequate overall approach for demonstrating biosimilarity.
3.4 Results of Clinical Comparability Studies
Clinical data are presented as raw data in Online Resource 2–5 (the product rows are not in the same order as Online Resource 1 to maintain anonymity). Table 3 provides a summary on all the uncertainties in clinical data, and how these were resolved.
3.4.1 PK Studies3.4.1.1 Rituximab
For all rituximab biosimilars, PK studies were performed in patients with rheumatoid arthritis (RA) with supportive PK data from oncology patients as part of the efficacy studies. With regard to the withdrawn rituximab application, a comparative efficacy study (in RA) that included PK similarity as a secondary objective was conducted prior to a dedicated comparative PK study [in non-Hodgkin’s lymphoma (NHL)] [43]. Length of follow-up ranged from 24 to 25 weeks (26 weeks in case of the withdrawn application). Primary endpoints [area under the curve to infinity (AUCinf), maximum concentration (Cmax) and AUC from time of administration up to the time of the last quantifiable concentration (AUClast) were contained within the pre-specified acceptance range for all approved biosimilars and secondary endpoints supported biosimilarity. Although for the withdrawn rituximab the pre-defined equivalence margin was 70-143% in the PK comparability study, the 90% CIs of the primary endpoints also met the standard equivalence margin of 0.8–1.25. As seen in Table 3, in two cases [26, 27, 53, 54, 65], results of a secondary endpoint were found outside of the standard acceptance limits, but deviations were seen as minor and not clinically relevant. Detailed information on the PK studies is available in Online Resource 2.
3.4.1.2 Trastuzumab
For all trastuzumab biosimilar candidates, PK studies were performed in healthy subjects with supportive PK data obtained in clinical trials in oncology patients. For the withdrawn trastuzumab application, an additional PK similarity study in healthy subjects was submitted. Length of follow-up of the PK studies ranged from 56 to 99 days (53 days in case of the withdrawn application). In all cases, the primary endpoints (AUCinf, Cmax and AUClast) were contained within the pre-specified acceptance range and secondary endpoints supported biosimilarity. Detailed information on the PK studies is available in Online Resource 4.
3.4.1.3 Population PK
Population PK (PopPK) was performed for some products, using different approaches [28, 37,38,39, 49]. Absence of PopPK analysis was accepted where PK similarity had been demonstrated in the dedicated PK study and PopPK was seen as supportive in the other cases.
3.4.2 Clinical Efficacy Studies3.4.2.1 Rituximab
Rituximab is currently approved in seven indications, both autoimmune and oncological [66]. Two applicants, including the one for the withdrawn rituximab MAA [27, 43, 53, 65], chose to compare efficacy in RA subjects as a model indication and the remaining two [26, 28, 54] chose follicular lymphoma (FL). Length of follow-up was up to 3 years. Overall response rate (ORR) was chosen as primary endpoint in FL and disease activity score using 28 joint counts (DAS28) or American College of Rheumatology Response (ACR 20) for RA. Detailed information on the efficacy studies is available in Online Resource 3.
3.4.2.2 Trastuzumab
Trastuzumab is currently approved in three indications [67]. For three biosimilars [37,38,39] metastatic breast cancer (MBC) was chosen as model indication in the pivotal clinical trial and for the remaining four, including the withdrawn MAA [36, 44, 48, 49], early breast cancer (EBC) was used. Length of follow-up was up to 3 years.
Three applicants chose ORR and the remaining four pathologic complete response (pCR) as the primary endpoint. Pre-specified equivalence margins for risk difference (RD) varied even though patient populations were the same as different reference studies were used for clinical and statistical justifications [37, 39, 48, 49]. Detailed information on the efficacy studies is available in Online Resource 5.
Table 3 shows those instances where some differences were found and how the remaining uncertainties were resolved. For two products [48, 49], the 95% CI of the difference in the pCR rates between treatments was not fully contained within the pre-defined equivalence margin, thus superiority of the biosimilar cannot be excluded.
3.4.2.3 Safety and Immunogenicity
The overall safety and immunogenicity profiles were compared descriptively and appeared similar between the biosimilars and the RP, as reviewed in detail by Kurki et al. [68].
With regard to the withdrawn rituximab biosimilar candidate application [43], the overall safety profile appeared to be similar in patients with RA but imbalances in adverse events (AEs), serious adverse events (SAEs), severity and deaths were observed in the comparative PK study in patients with NHL. Eight patients died in the product arm versus none in the reference arm; investigators assessed the causal relationship as not (6/8) or unlikely (2/8) related to study drug for all fatal SAEs.

Comments (0)