General information
The reagents and solvents used in this study were purchased from various chemical vendors such as Sigma-Aldrich (Pty) Ltd. (Johannesburg, South Africa), Merck (Pty) Ltd. (Johannesburg, South Africa). Reactions were monitored by thin layer chromatography (TLC) using Merck 60F254 silica gel plates (Merck, Johannesburg, South Africa) supported on aluminium sheets. Developed TLC plates were visualized under ultraviolet (UV254 and 366 nm) light or stained with iodine vapour. 1H and 13C NMR spectra were recorded on a Bruker Biospin 600 MHz spectrometer. The chemical shifts are given in values referenced to deuterated dimethylsulfoxide (DMSO-d6) and are reported in parts per million (ppm). Chemical shifts for DMSO-d6 appear at 2.5 ppm for 1H and 39.5 ppm for 13C. Proton coupling patterns are abbreviated as follows: s (singlet), d (doublet), dd (doublet of doublet), t (triplet), and m (multiplet). Coupling constants (J) are reported in Hz.
NMR data were analysed using MestReNova Software, version 5.3.2e4936. Melting points (mp) were established with a Büchi melting point B-545 instrument and were uncorrected. The high-resolution mass spectra (HRMS) were recorded by means of a Bruker micrOTOF-Q II mass spectrometer using atmospheric pressure chemical ionization (APCI) in positive ion mode. Infrared spectroscopy (FTIR) was determined with a Bruker ALPHA FTIR spectrometer.
General synthetic procedureSynthesis of intermediate 2
Into a round bottom flask containing 15 mL of ethanol, appropriate aniline (5 g), diethyl-2-(ethoxymethylene) malonate (1.5 equivalence) were added, and the mixture was refluxed for 24 h. Upon completion as indicated by TLC, it was allowed to cool at room temperature, washed with petroleum ether, filtered and dried. The dried crude was heated in Diphenyl ether at 240 °C for 1–2 h. Upon completion, the mixture was allowed to cool at room temperature and petroleum ether added to it. The resulting precipitate was washed with petroleum ether, filtered and dried to obtain intermediate 2 in 90% yield.
Synthesis of intermediated 3
Intermediate 2 (1 g) was treated with appropriate alkyl/aryl halide (2.5 eq) in THF:CHCl3 (2:1) and K2CO3 (1.5 eq) under refluxed for 24–36 h. Upon completion as indicated by TLC, the reaction mixture was placed in rotary evaporator for removal of solvent mixture. Water was added, and the resulting precipitate was filtered and dried to afford intermediate 3 [14].
Synthesis of compound 4–7 and 8
The N-alkylated intermediate 3 (0.5 g) was added with hydrazine (10 mL) and ethanol (5 mL) into a round bottom flask, the mixture was reflux for 24–36 h. Upon completion, crushed ice was added to mixture and left stirring for 30 min, after which the resulting precipitate was filtered and dried to obtain compounds 4–7. The same procedure was repeated with intermediate 2 to furnished compound 8.
Synthesis of compound 9–12
The hydrazide compound was stirred at room temperature in acetone for 1–2 h, filtered and dried to afford compound 9–12.
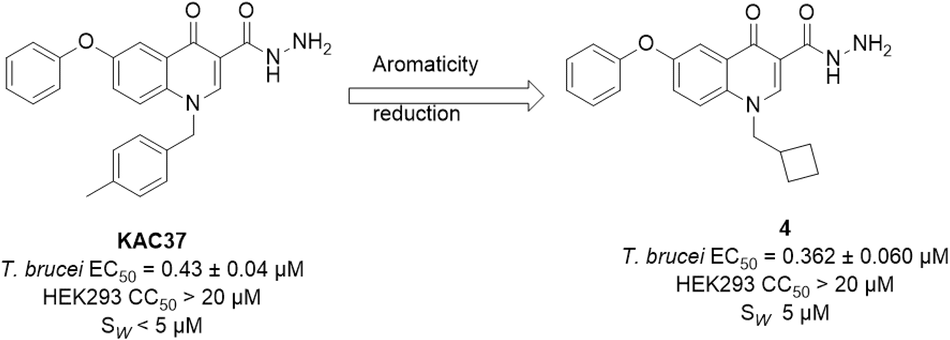
1-(Cyclobutylmethyl)-4-oxo-6-phenoxyquinolone-3-carbohydrazide,
4.
Brownish yellow powder, yields 59%, mp 200–204 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.57 (s, 1H), 8.84 (s, 1H), 7.98 (d, J = 9.3 Hz, 1H), 7.69 (d, J = 2.7 Hz, 1H), 7.59 (dd, J = 9.3, 2.7 Hz, 1H), 7.47 (t, J = 7.9 Hz, 2H), 7.25 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 7.9 Hz, 2H), 4.54 (d, J = 7.6 Hz, 4H), 2.82 (dd, J = 15.1, 7.5 Hz, 1H), 1.99–1.78 (m, 6H). 13C NMR (151 MHz, DMSO-d6) δ 174.55, 164.25, 156.32, 155.03, 147.24, 135.36, 130.77, 128.93, 124.88, 120.59, 119.98, 112.52, 110.07, 57.67, 34.61, 25.64, 18.15. HRMS APCl m/z calcd for C21H22N3O3 [M + H]+, 364.1656, found: 364.1670. IR (ATR) Vmax cm−1: 3648(N-H strectching), 3217(N-H stretching), 2993 (C-H stretching).
1-(Cyclopentylmethyl)-4-oxo-6-phenoxyquinolone-3-carbohydrazide,
5.
Brownish yellow powder, yields 60%, mp 313–316 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.84 (d, J = 9.9 Hz, 1H), 8.02 (t, J = 10.5 Hz, 1H), 7.70 (d, J = 2.7 Hz, 1H), 7.60 (dd, J = 9.2, 2.7 Hz, 1H), 7.51–7.44 (m, 2H), 7.25 (dd, J = 16.0, 8.6 Hz, 1H), 7.13 (t, J = 12.5 Hz, 2H), 4.56 (s, 2H), 4.45 (d, J = 7.5 Hz, 2H), 2.38 (dt, J = 15.1, 7.6 Hz, 1H), 1.72–1.57 (m, 4H), 1.50 (d, J = 4.7 Hz, 2H), 1.27 (dd, J = 21.8, 18.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 174.59, 164.26, 156.31, 155.06, 147.45, 135.31, 130.78, 128.95, 124.89, 120.64, 120.02, 112.54, 110.05, 57.74, 39.42, 29.94, 24.80. HRMS APCl m/z calcd for C22H24N3O3 [M + H]+, 378.1812, found: 378.1785. IR (ATR) Vmax cm−1: 3648 (N-H).
1-Benzyl-6-chloro-4-oxoquinolone-3-carbohydrazide,
6.
White powder, yields 87%, mp 379–381 °C. 1H NMR (600 MHz, DMSO) δ 11.39 (s, 1H), 10.22 (s, 2H), 9.33–9.02 (m, 1H), 8.41–8.16 (m, 1H), 7.99–7.62 (m, 2H), 7.57–7.07 (m, 5H), 6.06–5.69 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 173.90, 163.00, 149.55, 137.87, 135.36, 133.28, 130.60, 128.91, 128.27, 127.95, 126.45, 125.06, 120.74, 109.33, 56.18. HRMS APCl m/z calcd for C17H15ClN3O2 [M + H]+, 328.0847, found: 328.0857. IR (ATR) Vmax cm−1 : 3648 (N-H stretching), 2928 (C-H stretching).
1-(2-Hydroxyethyl)-4-oxo-6-phenoxyquinolone-3-carbohydrazide,
7.
Brown powder, yields 55%, mp 210–213 °C. 1H NMR (600 MHz, DMSO) δ 10.58 (d, J = 9.6 Hz, 1H), 8.74 (d, J = 3.9 Hz, 1H), 8.00 (t, J = 6.6 Hz, 1H), 7.77–7.67 (m, 1H), 7.63–7.54 (m, 1H), 7.52–7.43 (m, 2H), 7.31–7.21 (m, 1H), 7.13 (dd, J = 7.9, 4.2 Hz, 2H), 5.04–4.98 (m, 1H), 4.59–4.51 (m, 4H), 3.77 (dt, J = 8.8, 4.5 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 174.70, 164.33, 156.44, 154.89, 148.54, 135.49, 130.78, 128.99, 124.82, 120.50, 119.91, 112.67, 109.85, 59.16, 56.14. HRMS APCl m/z calcd for C18H18N3O4 [M + H]+, 340.1292, found: 340.1285. IR (ATR) Vmax cm−1: 3371(N-H stretching), 3262(C-H stretching), 3036 (O-H stretching).
4-Oxo-6-phenoxyquinolone-3-carbohydrazide,
8.
Brownish yellow powder, yields 72%, mp 336–339 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.92 (s, 1H), 10.63 (d, J = 11.2 Hz, 1H), 8.72 (d, J = 4.1 Hz, 1H), 7.78 (t, J = 6.3 Hz, 1H), 7.61 (d, J = 5.7 Hz, 1H), 7.56 (t, J = 7.2 Hz, 1H), 7.48–7.43 (m, 2H), 7.27–7.20 (m, 1H), 7.12 (dd, J = 8.0, 4.4 Hz, 2H), 4.55 (d, J = 11.2 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 175.25, 164.53, 156.59, 154.86, 143.02, 135.56, 130.73, 127.68, 125.26, 124.67, 121.88, 119.79, 111.95, 110.25. HRMS APCl m/z calcd for C16H14N3O3 [M + H]+, 296.1030, found: 296.1046. IR (ATR) Vmax cm−1, 3648(N-H stretching), 3254(N-H stretching), 3174(N-H stretching).
1-Ethyl-4-oxo-6-phenoxy-N’-(propan-2-ylidene)quinolone-3-carbohydrazide,
9.
White powder, yields 72%, mp 340–342 °C. 1H NMR (600 MHz, DMSO) δ 12.70 (d, J = 4.5 Hz, 1H), 8.95 (d, J = 2.9 Hz, 1H), 8.02 (t, J = 6.2 Hz, 1H), 7.73–7.68 (m, 1H), 7.68–7.62 (m, 1H), 7.53–7.45 (m, 2H), 7.30–7.23 (m, 1H), 7.16 (t, J = 5.9 Hz, 2H), 4.61–4.54 (m, 2H), 2.00 (d, J = 3.2 Hz, 3H), 1.93 (d, J = 3.1 Hz, 3H), 1.42 (dd, J = 10.0, 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 175.09, 160.76, 156.20, 155.37, 153.73, 147.89, 134.97, 130.84, 128.86, 125.21, 125.03, 120.52, 120.21, 112.33, 110.71, 49.18, 25.21, 17.79, 15.11. HRMS APCl m/z calcd for C21H22N3O3 [M + H]+,364.1654, found: 364.1647. IR(ATR) Vmax cm−1, 3309 (N-H stretching), 3174 (C-H stretching), 3050 (C-H stretching).
1-Benzyl-6-chloro-4-oxo-N’-(propan-2-ylidene)quinolone-3-carbohydrazide,
10.
White powder, yields 90%, mp 252–254 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.60 (s, 1H), 9.19 (s, 1H), 8.31 (s, 1H), 7.88–7.75 (m, 2H), 7.30–7.25 (m, 5H), 5.84 (s, 2H), 2.02 (d, J = 17.4 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 174.51, 159.79, 153.70, 149.46, 137.74, 135.48, 132.94, 130.23, 128.85, 128.18, 127.86, 126.40, 125.16, 120.46, 111.32, 56.07, 24.69, 17.18. HRMS APCl m/z calcd for C20H19ClN3O2 [M + H]+, 368.1160, found: 368.1164. IR (ATR) Vmax cm−1; 3648 (N-H stretching), 3040 (C = H stretching), 2913 (C-H stretching).
6-Chloro-1-(cyclopentylmethyl)-4-oxo-N’-(propan-2-ylidene)quinolone-3-carbohydrazide,
11.
White powder, yields 92%, mp 230–233 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 8.97 (s, 1H), 8.30 (s, 1H), 8.04 (d, J = 9.1 Hz, 1H), 7.87 (d, J = 8.7 Hz, 1H), 4.49 (d, J = 7.4 Hz, 2H), 2.36 (dd, J = 14.8, 7.3 Hz, 1H), 2.01 (d, J = 22.1 Hz, 6H), 1.64–1.28 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 174.24, 159.87, 153.53, 148.66, 137.57, 132.86, 130.14, 128.02, 125.10, 120.28, 110.74, 57.25, 38.89, 29.34, 24.67, 24.25, 17.17. HRMS APCl m/z calcd for C19H23ClN3O2 [M + H]+, 360.1473, found: 360.1457. IR (ATR) Vmax cm−1: 3648(N-H stretching), 2963 (C = H stretching), 2943 (C-H stretching).
1-Benzyl-6-bromo-4-oxo-N’-(propan-2-ylidene)quinolone-3-carbohydrazide,
12.
Cream white powder, yields 86%, mp 257–259 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 9.21 (s, 1H), 8.45 (s, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.74 (d, J = 9.1 Hz, 1H), 7.35 (t, J = 7.1 Hz, 2H), 7.30 (d, J = 6.9 Hz, 1H), 7.23 (d, J = 7.3 Hz, 2H), 5.84 (s, 2H), 2.01 (d, J = 18.5 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 174.40, 159.77, 153.72, 149.48, 138.06, 135.60, 135.47, 128.84, 128.40, 127.85, 126.38, 120.56, 118.33, 111.43, 56.02, 24.69, 17.17. HRMS APCl m/z calcd for C20H19BrN3O2 [M + H]+, 412.0633, found: 412.0655. IR (ATR) Vmax cm−1; 3566(N-H stretching), 3254 (C = H stretching), 2964(C-H stretching).
In vitro antitrypanosomal assay against T. b. brucei
Bloodstream forms of T. b. brucei Lister 427 were grown in HMI-9 medium 37 °C in 5% CO2. Parasites were maintained in exponential growth phase and passaged every 48–72 h. Growth inhibition of T. b. brucei was determined using a fluorescence-based resazurin cell viability assay. Test compounds were serially diluted in DMSO and added to 96-well polystyrene assay plates to give final assay concentrations ranging from 4 μM–0.87 nM (1 μL; 0.5% total DMSO). HMI-9 medium (99 μL/well) was added, followed by 2 × 105 parasites/mL of parasites in HMI-9 medium (100 μL) for a total density of 2 × 104 trypanosomes/well. Assay plates were incubated at 37 °C and 5% CO2 for 72 h, followed by addition of 20 μL 0.5 mM resazurin (Alfa Aesar, Cat. B21187) in PBS. Assay plates were incubated in the dark for 2–4 h at 37 °C. Fluorescence was measured using a 2104 EnVision® multilabel plate reader at 531 nm excitation and 595 nm emission wavelengths. All assays were normalized to positive and negative controls in each assay plate. Dose-response curves were generated and EC50 values calculated with GraphPad Prism, version 9.3 (San Diego, CA) using a sigmoidal four parameter logistic curve [15].
In vitro cytotoxicity evaluation
HEK293 cells were grown in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin. Cells were maintained at 37 °C in 5% CO2 and sub-cultured when at 60–80% cell confluence. Cytotoxicity was measured using a fluorescence-based resazurin cell viability assay. Test compounds were serially diluted in DMSO and added to 96-well polystyrene assay plates to give final assay concentrations ranging from 20 μM–0.87 nM (1 μL; 1% total DMSO). Fresh medium was added to the assay plate (49 μL/well), followed by 4 × 105 cells/mL solution of HEK293 cells in DMEM (50 μL) for a total density of 2 × 104 cells/well. Assay plates were incubated at 37 °C and 5% CO2 for 48 h, followed by addition of 20 μL 0.5 mM resazurin (Alfa Aesar, Cat. B21187) in PBS to each well. Assay plates were incubated in the dark for 2–4 h at 37 °C. Fluorescence was measured using a 2104 EnVision® multilabel plate reader at 531 nm excitation and 595 nm emission wavelengths All assays were normalized to positive and negative controls in each assay plate. Dose-response curves were generated and CC50 values calculated with GraphPad Prism, version 9.3 (San Diego, CA) using a sigmoidal four parameter logistic curve [10].
Solubility determination
Aqueous solubility was determined in 96-well plate format at pH 6.5 using the miniaturised shake-flask method reported previously [16]. Briefly, a 10Mm stock of the sample was prepared in DMSO, from which 4 μL was aliquoted to a 96-well plate and evaporated using a GeneVac system. Phosphate buffer saline (pH 6.5) was then added to the compounds containing wells, and the plate incubation for 24 h at 25 °C while shaking. The plates were centrifuged at 3500 g for 15 min, and then analysed using HPLC-DAD. For each sample, the calibration curve from which the solubility was determined was generated from a concentration range of 10–220 μM in DMSO [17].
Metabolic stability testing
Metabolic stability was determined using a previously reported procedure (Dube et al., 2023). Selected compounds at 1 μM concentration were incubated with human (mixed gender, Xenotech), rat (male IGS, Xenotech) and mouse (male CD1, Xenotech) liver microsomes (0.4 mg/mL) at 37 °C for 30 min, after which ice cold acetonitrile containing internal standard was added to quench the reaction. Samples were centrifuged and then analysed by LC–MS/MS for the disappearance of the parent compound. Half-life, clearance and hepatic excretion ratios were determined using standard equations, as previously reported [18].
Lipophilicity (LogD)
The lipophilicity of the compounds was measured using a miniaturised shake-flask method in a 96-well plate format (Kerns and Di 2008). Briefly, equal volumes of 1-octanol and phosphate buffer, pH 7.4, were added to 10 µM of each compound in a deep-well plate. The plate was shaken vigorously for 2 h at 25 °C. The phases were then carefully separated and transferred to another plate. Analysis was performed by HPLC-UV. LogD values were determined from the peak areas of the compound in octanol and buffer phases using the formula LogD7.4 = log (Aoctanol\Abuffer), where A is the peak area [17].


Comments (0)