
Remember me
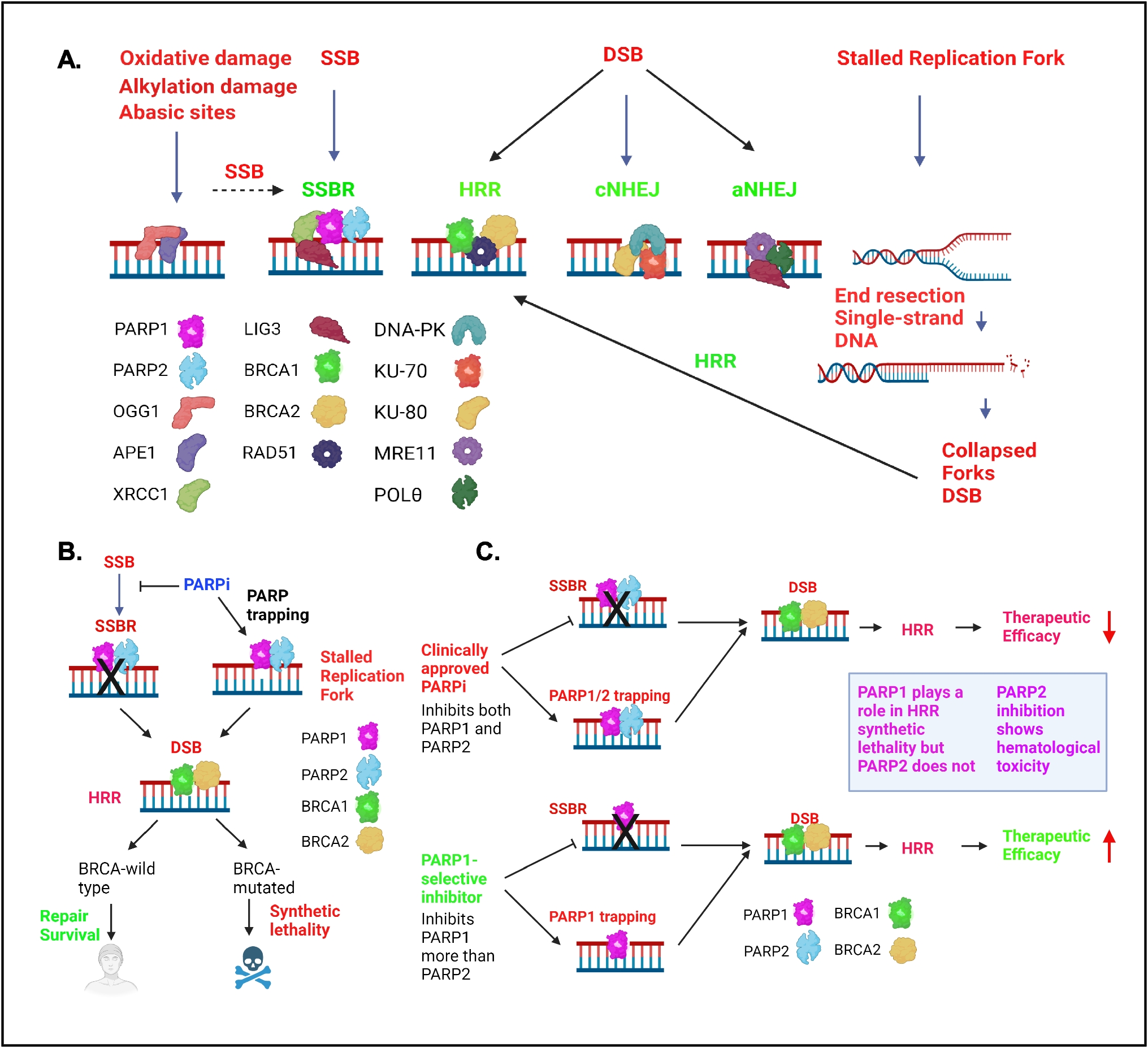


Poly (ADP‒ribose) polymerase (PARP) plays a pivotal role in cellular physiology by catalyzing the transfer of ADP‒ribose from nicotinamide (NAD +) precursors to target proteins, a posttranslational modification called PARylation [1, 2]. This enzymatic process, termed poly (ADP)-ribosylation, regulates vital cellular pathways [2, 3]. PARP1 and PARP2 are the best characterized PARPs (which refers to PARP 1 and 2) and contribute to most PARP activities [4,5,6]. PARP1 and PARP2 are well characterized for their role in DNA damage repair, ensuring cell survival by modifying proteins involved in chromatin architecture and DNA repair processes [5, 7, 8]. PARP1 has the predominant role in DNA damage repair and PARP2 has a lesser effect [9]. PARP is catalytically activated by single-strand breaks (SSBs) and double-strand breaks (DSBs) [5, 10,11,12,13]. Subsequently, PARP promotes the formation of poly (ADP‒ribose) (PAR) chains, which are important for providing a docking platform for DNA repair proteins to damaged sites, facilitating their recruitment [1, 10, 12]. Finally, the PAR chains are removed from substrate proteins by dePARylating enzymes [14, 15]. Recently, a variety of ADP-ribose degrading enzymes have been identified with different substrate specificities. Poly (ADP-ribose) glycohydrolase (PARG) is the major dePARylating enzyme which hydrolyzes the glycosidic linkages between ADP-ribose units of PAR polymers to generate free ADP-ribose monomers [14, 15]. Recently, other dePARylating enzymes such as ADP-ribose hydrolases, phosphodiester ADP-ribose hydrolases, macrodomain-ADP-ribose erasers, have been identified which are also involved in removing PAR chains (reviewed in [16]. Owing to the catalytic function of PARP, PARP can tightly bind to DNA, resulting in “PARP-DNA” trapping, which is stabilized by PARP inhibitors (PARPis), blocking the replication machinery and facilitating the collapse of the replication fork [17]. Disruption of these processes by pharmacological inhibition has emerged as a promising strategy in cancer therapy.
PARP Plays a Role in Base Excision Repair/Single-Strand Break Repair and DNA ReplicationDue to spontaneous DNA damage, oxidative stress, irradiation, and reactive endogenous metabolites, thousands of DNA lesions are introduced in the DNA every day. These DNA modifications are repaired by base excision repair (BER) and single-strand break repair (SSBR). BER is the major pathway for repairing oxidative base damage, alkylation damage, and abasic sites [18]. In BER, damage is detected by OGG1 and APE1, which create SSBs that are repaired by the SSBR pathway [5, 13] (Fig. 1A). PARP1/2 binds to SSBs with high affinity and becomes activated, inducing ADP-ribosylation of histones and other DNA repair proteins and essential for BER/SSBR pathways [5, 19]. The recruitment of the core factor XRCC1 to SSBs is dependent on PARP1 and PARP2 [20]. XRCC1 functions as a scaffold for DNA Polβ, LIG1/3 and PNKP needed for the repair process [5, 18] (Fig. 1). Thus, BER/SSBR restores the damaged DNA to intact DNA [8].
Fig. 1
Major cellular DNA damage repair pathways related to PARP function and the mechanism of synthetic lethality between PARP inhibitors and HRR deficiency. A. PARP1/2 predominantly play a role in SSBR. Inhibition of SSBR results in DSBs. PARP inhibition and PARP trapping result in stalled replication forks, which are converted to DSBs. In the presence of BRCA1/2, DSBs are predominantly repaired by HRR. In the absence of BRCA1/2, cells repair DSBs via the cNHEJ and aNHEJ pathways. BER (base excision repair), SSBR (single-strand break repair), HRR (homologous recombination repair), cNHEJ (classical nonhomologous end joining), aNHEJ (alternative nonhomologous end joining). B. Schematic showing the synthetic lethality between a PARPi and HRR-deficiency. C. Schematic showing how PARP1-selective inhibitors improve PARP efficacy. PARP1 is needed for the synthetic lethality of HRR and PARP1 selective inhibitors limit toxicity
PARP1 interacts and stimulates the activity of multiple replication proteins and influences DNA replication process [21,22,23,24]. Replication stress caused by oncogene activation, dNTP depletion, and unrepaired SSBs. These SSBs encounter the replication machinery resulting in stalled and/or collapsed replication fork with a single-ended DNA DSBs [8]. PARP1 has been implicated in protecting the degradation of stalled DNA replication forks and DSBs by promoting the recruitment of MRE11 and RAD51 [5, 25, 26]. These DSBs are resolved by the homologous recombination repair (HRR) pathway (described further below). PARP1 also plays a role in the prevention of untimely restart of stalled replication forks by binding to RECQ1 and allowing repair of DNA lesions [5]. Recently, PARP1 has been implicated to play a direct role in the Okazaki fragment processing [27, 28]. These studies showed that the unligated Okazaki fragments is one of the major sources of PARP activity and perturbation of the DNA replication proteins LIG1 or FEN1 increases the S phase PARP independent of damaged DNA or replication stress [27, 28].
PARP Inhibitors Inhibit Single-Strand Break Repair, DNA Replication, and induce PARP Trapping Resulting in Double-Strand Breaks which are Repaired by Homologous Recombination Repair PathwayPARPis catalytically inhibit PARP1/2, and the Inhibition of SSBR by PARPi results in accumulation of SSBs. When these SSBs are unrepaired and enter the S phase, they are converted to more lethal DSBs. Furthermore, upon PARPi treatment, PARP binding to the DNA breaks is stabilized resulting in PARP trapping (Fig. 1A) [17, 29]. These “trapped-PARPs” causes replication fork stalling and SSBs which eventually gets converted to DSBs (Fig. 1A) [17, 30,31,32,33] (Fig. 1A). Studies supported that trapped PARPs form a complex with damaged DNA which is more cytotoxic compared with unrepaired SSBs due to PARP inhibition [17]. In cells harboring wild-type BRCA1/2 genes, these DSBs are mainly repaired via the HRR pathway, an error-free repair pathway which predominantly works in S phase and to a lesser extent in G2 phase of cell cycle due to availability of the homologous chromatid for recombination (Fig. 1A) [8, 34,35,36]. In the canonical HRR, BRCA2 initially binds to RAD51 and localizes to the DSB sites, whereas BRCA1 plays a crucial role in DSB resection and signal transduction [37, 38]. BRCA1 interacts with PALB2 and helps localization of BRCA2 by forming a molecular scaffold of BRCA1-PALB2-BRCA2 needed for HRR [39]. In the absence of functional BRCA1 and BRCA2, DSBs are resolved through the error-prone classical nonhomologous end joining (cNHEJ) pathway, or the alternative nonhomologous end joining (aNHEJ) pathway (Fig. 1A). During cNHEJ process, DNA-PKcs, KU-70, and KU80 bind the DSBs. 53BP1 limits end resection to create an overhang to promote ligation by DNA ligase 4 and XRCC4-XLF [5]. During the aNHEJ, DSBs use microhomologies to align the broken DNA ends. MRE11, POLꝊ and other proteins bind to the microhomology region and LIG3 play a role in the ligation of broken DNA ends (Fig. 1A) [8, 32, 40]. The ligation of broken DNA ends often lead to genomic alterations and increased heterogeneity in the genome [8, 32, 36, 40].
PARP Inhibitors Exhibit Synthetic Lethality in BRCA-Mutated CancersAs described in the previous section, the unrepaired SSBs, defective replication, and PARP trapping induce replication stress-induced DSBs. BRCA-wild-type cells repair these DSBs via the HRR pathway [37, 38], but the BRCA-mutated cells cannot repair these DSBs. Therefore, inhibition of PARP in BRCA-mutated cells causes synthetic lethality due to the presence of unrepaired DSBs [41,42,43,44,45,46] (Fig. 1B). As a result, BRCA1/2-mutated cells are hyper reliant on PARP-mediated DNA repair for their survival [31, 41, 42]. This laid the foundation for the development of targeted therapies by exploiting the vulnerability of BRCA1/2-mutated tumors to homologous recombination deficiency (HRD) [47]. Currently, HRD is used as a predictive biomarker for PARP treatment and several methods are being used to determine the level of HRD in cancers [8].
PARP Inhibitors Exhibit Synthetic Lethality in Patients with the Brcaness PhenotypeNumerous investigations have shown that in addition to classical BRCA1 and BRCA2 mutations, a diverse array of molecular alterations can contribute to the development of HRD [40, 48,49,50]. Defects in these non-BRCA mutated proteins can promote a"BRCAness"phenotype, characterized by biological and clinical features akin to those associated with BRCA1/2 mutations [40, 48,49,50]. These alterations include mutations in key genes that play a role in HRR, such as RAD50, RAD51, RAD51C, RAD51D, RAD54L, and RAD51C58, as well as the overexpression of the HORMAD1 protein, which suppresses RAD51-dependent HRR [48, 49, 51, 52]. Additionally, defects in DNA damage response genes associated with HRR, such as BARD1, BRIP1, ATM, ATR, CHK1, CHK2, NBS1, PRKDC, RPA1, and DSS1, and mutations in several Fanconi anemia pathway genes, including PALB2, FANCA, FANCC, FANCD2, FANCE, and FANCF, have been implicated in the development of HRD [46, 48, 53]. Moreover, mutations in non-HRR DNA damage response genes such as loss of CDK12 can induce genomic instability resembling BRCAness across various cancer types, indirectly promoting HRR deficiency [54, 55]. CRISPR-Cas9 loss-of-function screens in PARPi-insensitive cell lines has identified most frequently inactivated genes in tumors and showed that XRCC1 is a potential new prognostic biomarker of PARPi sensitivity in prostate cancer [56].
Other mechanisms, including promoter methylation, somatic BRCA1/2 mutation, and gene deletion, can also impair BRCA1/2 function, resulting in a BRCAness phenotype [57,58,59]. PARPi induce synthetic lethality not only in patients harboring hereditary BRCA1/2 mutations but also in those exhibiting the BRCAness phenotype and HRD [41, 54]. Considering all these factors, HRR gene mutations beyond BRCA1/2 and overall dysregulation of HDR can serve as biomarkers to predict sensitivity to PARPis, thereby guiding personalized therapeutic strategies [48].
First-Generation PARP Inhibitors Targeting both PARP1 and PARP2Multiple PARPis have undergone successful clinical trials and have been approved by the Food and Drug Administration for ovarian, breast, pancreatic, and prostate cancers, demonstrating the promise of this approach [31, 60, 61]. These PARPis exhibit varying potencies as both PARP catalytic function inhibitors and PARP trapping agents and have been reviewed elsewhere [17, 31, 62, 63]. In general, the first-generation PARPis olaparib, talazoparib, niraparib, rucaparib, veliparib, pamiparib, and fuzuloparib target both PARP1 and PARP2, bind to their active sites and inhibit PARylation. This is because PARP1 and PARP2 share a high degree of sequence homology (69%) at the catalytic domain [64]. Even though all these inhibitors trap PARP at sites of DNA damage they show varied efficacy in PARP trapping [30, 61]. Compared with other agents, talazoparib results in the greatest amount of PARP trapping, in contrast with veliparib, which has weaker PARP trapping activity [30]. Several clinical trials have extensively explored the clinical efficacy of olaparib [65], talazoparib [66, 67], niraparib [68], rucaparib [
Comments (0)