
Remember me
Major depressive disorder (MDD) is a common psychiatric condition with a rising prevalence and a substantial impact on functioning.1 Although treatment can improve quality of life, many patients still experience lower quality of life than the general population, even among patients who achieve remission.2 Patients with MDD have a significantly higher risk of comorbidities, including metabolic syndrome, obesity, and cardiovascular disease, which can increase illness burden.3
Antidepressant therapies (ADTs), including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors, are limited by delayed responses and undesirable side effects (eg, weight gain, metabolic disturbances, sexual dysfunction).4–6 Available treatments lead to ≈25% and ≈40% of patients achieving remission or response, respectively, following first-line treatment as reported in the STAR*D trial reanalysis.7
Approximately 50% of patients with MDD have inadequate ADT response, defined as failing to achieve ≥50% improvement in depression severity after ≥6–8 weeks of treatment.8 One in 6 patients with inadequate ADT response reports that poor efficacy and tolerability issues contribute to reduced medication adherence.8 Patients with inadequate response have significantly impaired functioning and greater emergency room utilization/hospitalization compared with patients who respond.8 To treat MDD in patients with inadequate response, augmenting ADT treatment with atypical antipsychotics has received increased attention in recent years.9 Aripiprazole, quetiapine extended release, brexpiprazole, and cariprazine are US Food and Drug Administration (FDA)–approved for adjunctive treatment to ADT in MDD.10,11 Because some antipsychotics adjunctive to ADT have reported safety concerns (eg, extrapyramidal symptoms [EPS], metabolic effects, prolactin increase, weight gain),6,12,13 a novel combination of adjunctive antipsychotic with ADT resulting in tolerable safety and improved efficacy is needed.
Lumateperone, a mechanistically novel antipsychotic, is US FDA approved in adults to treat schizophrenia and depressive episodes associated with bipolar I or bipolar II disorder as monotherapy and as adjunctive therapy with lithium or valproate.14,15 Lumateperone is a potent serotonin 5-HT2A receptor antagonist, a dopamine D2 receptor presynaptic partial agonist and postsynaptic antagonist, a D1 receptor–dependent indirect modulator of glutamatergic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDA (N-methyl-D-aspartate) currents, and a serotonin reuptake inhibitor (SRI).15,16 Lumateperone has negligible binding to H1, 5-HT2c, and muscarinic receptors, which may limit weight gain, and has lower striatal D2 receptor occupancy compared with other antipsychotics, which may reduce EPS risk.15,17 SRIs are known anxiolytics, and 5-HT2A antagonists can enhance the anxiolytic effects of SSRIs; additionally, preclinical studies indicate NMDA modulation may also provide anxiolytic activity.18,19 This novel mechanism of action, in part targeting the glutamatergic system, may yield particular efficacy in MDD due to the involvement of glutamate in depression.20
This phase 3, randomized, double-blind, placebo-controlled trial investigated efficacy and safety of adjunctive lumateperone 42 mg in patients with MDD with inadequate ADT response.
METHODS Trial OversightThe sponsor (Intra-Cellular Therapies, a Johnson & Johnson company) designed and oversaw conduct of the trial and data analysis. The study was approved by the appropriate institutional review board/independent ethics committee and performed in accordance with the Declaration of Helsinki, in compliance with Good Clinical Practice guidelines. Written informed consent was obtained from each patient before entering the study, following the explanation of the study procedures and possible side effects. The authors vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol. All the authors contributed to writing the manuscript (assisted by a sponsor-funded medical writer) and to the decision to submit the manuscript for publication.
PatientsEligible adults (18–65 years, inclusive) met Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5),21 criteria for MDD, confirmed using the Mini-International Neuropsychiatric Interview.22 Patients had inadequate response to 1 or 2 ADTs during the current major depressive episode (MDE), per the Antidepressant Treatment Response Questionnaire23 (defined as <50% improvement with ≥6-week ADT with adequate treatment of at least the minimum effective dose per package insert). Patients were experiencing an MDE beginning ≥8 weeks and ≤18 months before screening. At screening and baseline, patients had Montgomery-Åsberg Depression Rating Scale (MADRS)24 Total score ≥24, Clinical Global Impression Scale-Severity (CGI-S)25 score ≥4, and Quick Inventory of Depressive Symptomatology-Self Report-16 item (QIDS-SR-16)26 score ≥14.
Patients were excluded if there was a ≥25% decrease in MADRS Total or QIDS-SR-16 Total scores between screening and baseline, a significant risk for suicidal behavior, or a lifetime history of treatment resistance (no remission) to ≥3 approved treatments at the adequate dose for an adequate duration. Full eligibility criteria are available in the Supplementary Methods.
Study DesignThis phase 3, randomized, double-blind, placebo-controlled, parallel-group, fixed-dose study (ClinicalTrials.gov, NCT04985942) was conducted from July 30, 2021 to February 27, 2024, across 54 sites in the US, Bulgaria, Czechia, Hungary, India, and Slovakia. The study included a 2-week screening period, 6-week double-blind treatment period, and 1-week safety follow-up period. Patients were randomized 1:1 at baseline via an interactive web response system to receive adjunctive placebo or adjunctive lumateperone 42 mg. Capsules were identical in appearance, provided in a blister card at weekly visits, and self-administered orally once daily in the evening. Treatment compliance was assessed by the proportion of capsules remaining in the blister pack at each visit. Efficacy and safety assessments occurred weekly on day 1 and days (±3 days) 8, 15, 22, 29, 36, and 43, and safety follow-up occurred on day 50 ±3 days.
AssessmentsThe primary and key secondary efficacy outcomes were change from baseline to day 43 in MADRS Total score (range from 0 to 60; higher scores indicating more severe depression) and CGI-S score (range from 1 to 7; higher scores indicating greater severity), respectively. Change from baseline to day 43 in patient-rated scales for depression (QIDS-SR-16 Total score, range from 0 to 27; higher scores indicating more severe depression) and anxiety (Generalized Anxiety Disorder-7 [GAD-7] Total score, range 0–21; higher scores indicating more severe anxiety),27 and response (≥50% MADRS Total score decrease from baseline to day 43) and remission (MADRS Total score ≤10 at day 43) status were assessed. MADRS Total score and CGI-S score were also evaluated in patients by number of treatment failures and in patients meeting DSM-5 anxious distress criteria (Supplementary Methods).
Safety assessments included adverse events (AEs), EPS, suicidality, clinical laboratory, vital sign, and electrocardiogram measurements. Suicidality was evaluated by the Columbia-Suicide Severity Rating Scale (C-SSRS)28 and treatment-emergent AEs (TEAEs). EPS were assessed with the Abnormal Involuntary Movement Scale (AIMS),25 Barnes Akathisia Rating Scale (BARS),29 and Simpson Angus Scale (SAS)30 and by TEAEs. AEs were evaluated using the Medical Dictionary for Regulatory Activities (MedDRA) version 24.0.
Statistical AnalysesPrimary and key secondary efficacy analyses were performed using the hypothetical estimand strategy, which assumed patients were on study treatment up to day 43 and efficacy data collected after patients discontinued study treatment or initiated new ADT were not included. The primary efficacy analyses used a mixed-effects model for repeated measures (MMRM), including the change from baseline in MADRS Total score at each study visit as the response variable, and treatment group, study visit, site (or pooled site) as factors and the baseline MADRS Total score as covariate, and interaction terms for baseline MADRS Total score-by-study visit and treatment group-by-study visit. An unstructured covariance matrix was used to estimate the correlation among repeated measurements within patient. The mean treatment difference for lumateperone vs placebo was estimated via contrast based on treatment group and treatment group-by-study visit factors. Hypothesis test was performed at the 2-sided 5% significance level; CIs were 2-sided 95% CIs. A fixed sequence testing procedure was used to control the overall Type I error of 0.05 with the primary end point based on the primary estimand approach; if significant, the secondary end point was tested at 0.05. The key secondary efficacy end point was analyzed similarly to the primary end point.
MADRS response and remission statuses were evaluated using logistic regression with terms for treatment group and baseline MADRS Total score, with patients with missing MADRS Total score considered as nonresponders or nonremitters. The change from baseline at day 43 in QIDS-SR-16 Total score was evaluated by an analysis of covariance model with terms for treatment group, site (or pooled site), and baseline total score. The change from baseline at day 43 in GAD-7 Total score was analyzed using an MMRM similar to that used in the primary end point analyses. Efficacy analyses were evaluated in the modified intent-to-treat (mITT) population, defined as all randomized patients who received ≥1 treatment dose, had a baseline MADRS Total score, and had ≥1 postbaseline MADRS Total score. Patient-reported outcomes (QIDS-SR-16 and GAD-7 Total scores) were analyzed in the intent-to-treat population, defined as all randomized patients who received ≥1 treatment dose and had a baseline MADRS Total score. Demographic and safety parameters were summarized descriptively in all randomized patients who received ≥1 treatment dose. Additional details are provided in the Supplement. Data within the manuscript have been reported in accordance with 2010 CONSORT guidelines.
RESULTS Patient PopulationOf 700 screened patients with MDD assessed for eligibility, 485 were randomized to receive treatment (placebo + ADT, 243; lumateperone + ADT, 242; Supplementary Figure 1). The safety and mITT populations comprised 484 and 481 patients, respectively. Most patients (placebo + ADT, 95.5%; lumateperone + ADT, 91.3%) completed double-blind treatment. Overall, the most common reasons for treatment discontinuation in the safety population were AEs (3.3%) and withdrawal of consent (1.9%). Mean treatment compliance was >99% in both treatment groups.
Baseline demographics and characteristics were similar between groups (Table 1). Most patients were female (65.7%) and White (76.7%). The most common ADT was citalopram/escitalopram (placebo + ADT, 35.4%; lumateperone + ADT, 34.9%). Both treatment groups had moderate-to-marked depression per MADRS Total and CGI-S scores.31
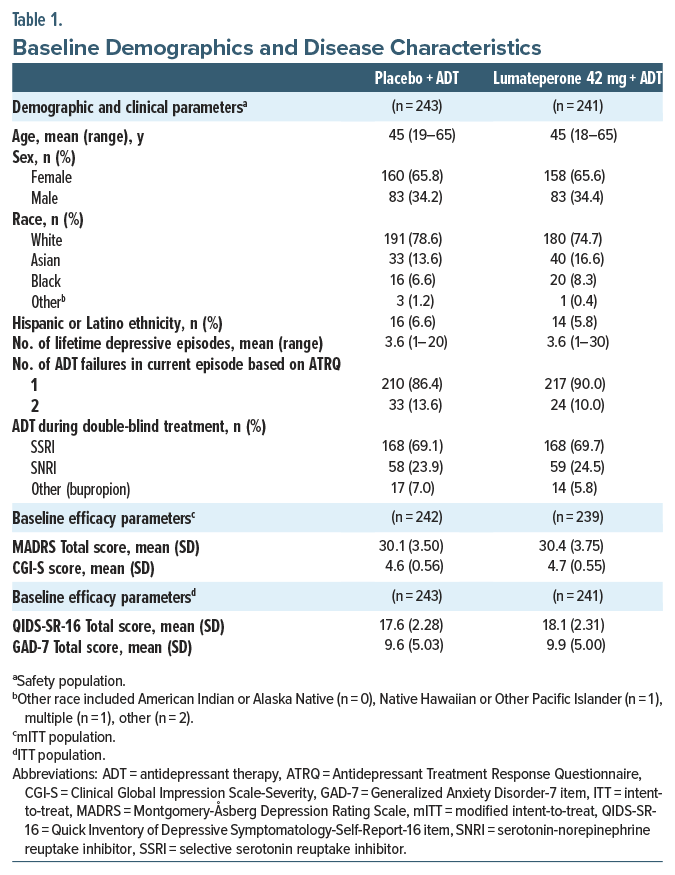
Lumateperone + ADT met the primary end point, significantly reducing MADRS Total score from baseline to day 43 vs placebo + ADT (least squares mean difference [LSMD] vs placebo=−4.9; 95% CI, −6.38 to −3.44; effect size [ES]=−0.61; P <.0001; Figure 1A, Table 2). Additionally, MADRS response and remission rates at day 43 were significantly greater with lumateperone + ADT than placebo + ADT (Table 2). Lumateperone + ADT also met the key secondary end point, with significant reduction in CGI-S score from baseline to day 43 vs placebo + ADT (LSMD=−0.7; 95% CI, −0.85 to −0.48; ES =−0.67; P <.0001; Figure 1B, Table 2). Lumateperone + ADT treatment significantly improved patient-reported depression and anxiety severity from baseline to day 43 vs placebo + ADT, measured by QIDS-SR-16 and GAD-7 scores, respectively (Figure 1C, Table 2).
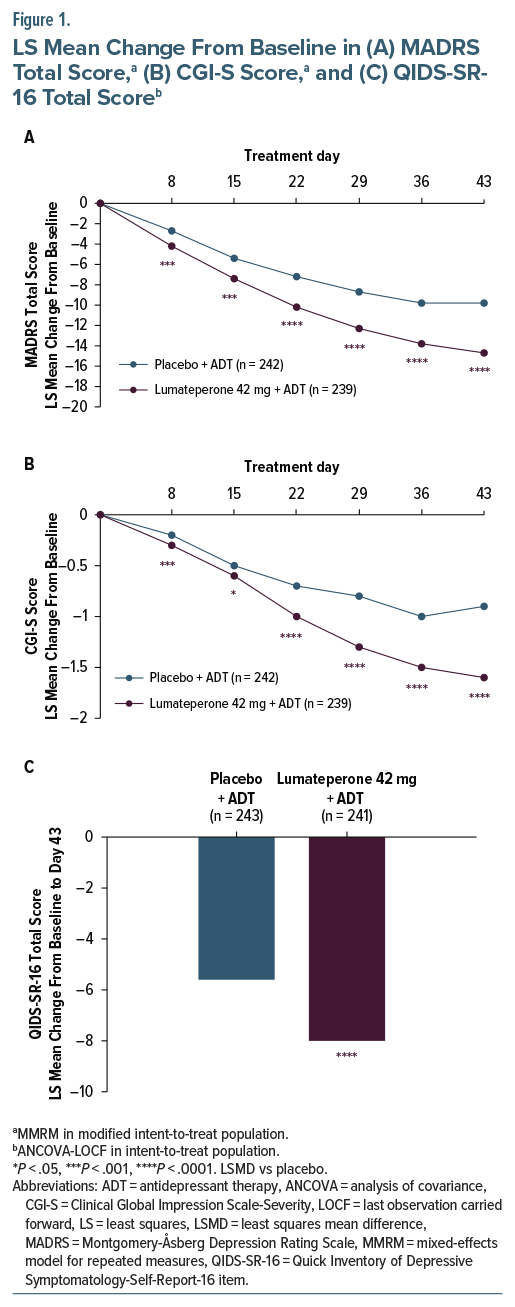
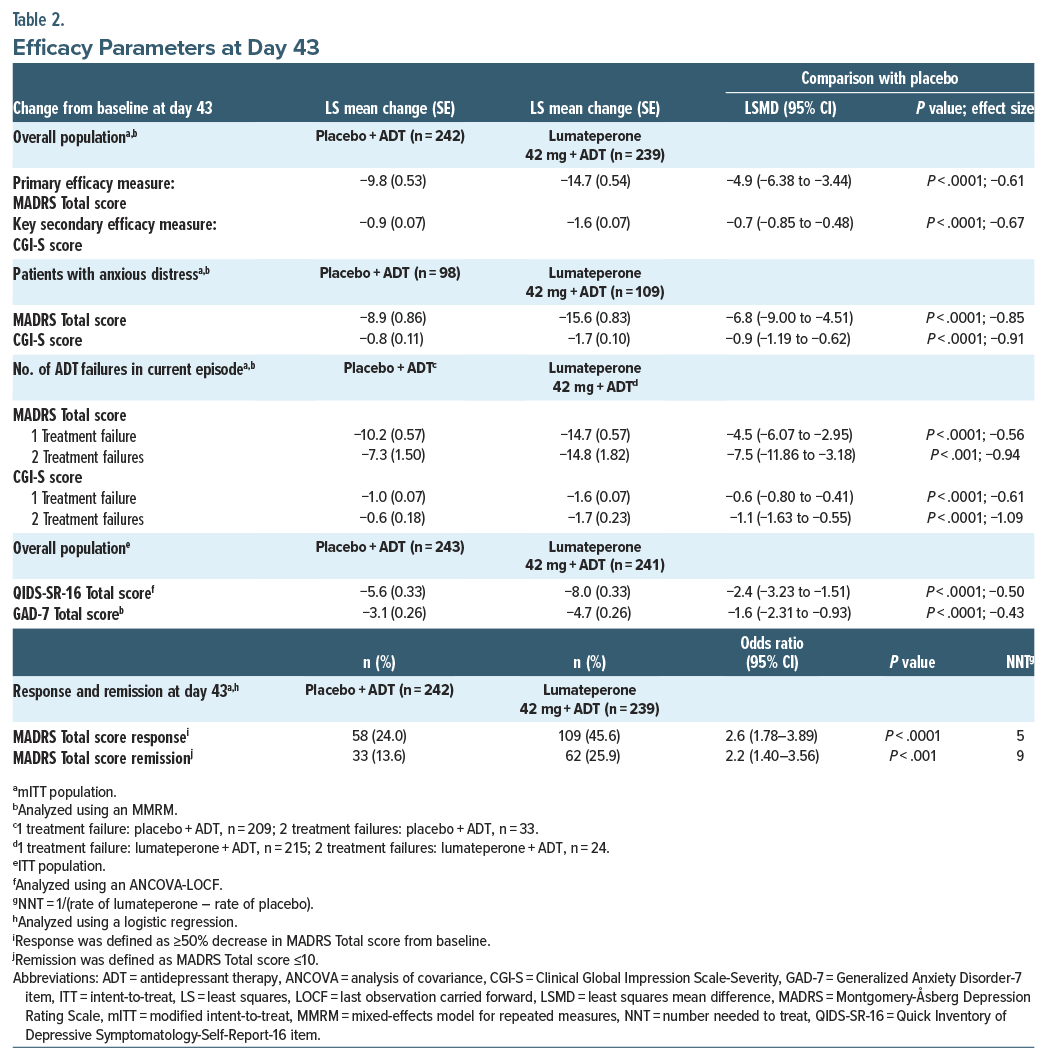
DSM-5 anxious distress criteria (Supplementary Methods) were met by nearly half of patients in the safety population at baseline (placebo + ADT, n=99 [40.7%]; lumateperone + ADT, n=110 [45.6%]). In patients with MDD and anxious distress, lumateperone + ADT significantly improved MADRS Total score and CGI-S score from baseline to day 43 (Table 2). Additionally, lumateperone + ADT treatment significantly improved both MADRS Total score and CGI-S score from baseline to day 43 in patients with 1 or 2 treatment failures (Table 2).
SafetyTEAEs occurred in 46.5% and 58.1% of the placebo + ADT and lumateperone + ADT groups, respectively (Table 3). The most common TEAEs (≥5%, more than twice placebo + ADT) were dry mouth, fatigue, and tremor. Most TEAEs (>98%) were mild or moderate severity. Of the 2 patients receiving placebo + ADT and 13 patients receiving lumateperone + ADT who discontinued treatment due to a TEAE, the only TEAEs associated with >1 patient discontinuing in either treatment group were fatigue (n=3), dizziness (n=3), and sedation (n=2) in the lumateperone + ADT group (Supplementary Table 1). All TEAEs leading to treatment discontinuation had resolved at follow-up. No patients died during the study.
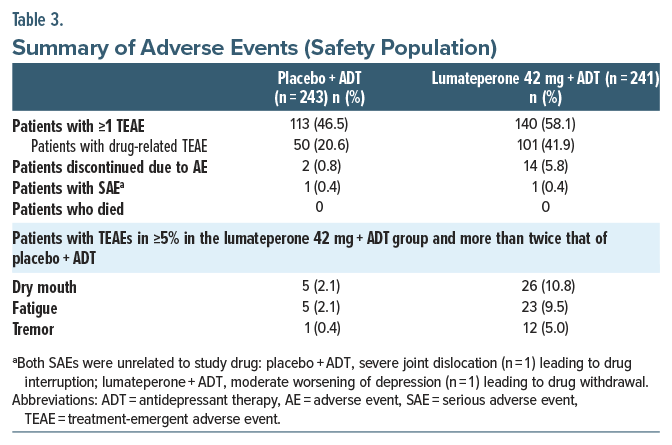
No notable changes occurred in EPS-related scales, including AIMS, BARS, and SAS Total scores (Supplementary Table 2). The incidence of akathisia, based on shift from baseline in BARS score, and incidence of Parkinsonism, based on shift in SAS score, were low during treatment (Supplementary Table 2). According to broad standard MedDRA query, EPS-related TEAEs occurred in 7 patients (2.9%) in the placebo + ADT group and 15 patients (6.2%) in the lumateperone + ADT group. Among the EPS-related TEAEs, the tremor was observed in 12 patients in the lumateperone + ADT group and in 1 patient in the placebo + ADT group. Among the patients in the lumateperone + ADT group, none were severe in intensity, and most were mild (9 patients) or moderate (3 patients). The onset of most of the tremors occurred during the first 2 weeks of the treatment period. Mean (SD) duration was 14.6 days (±10.65 days). Of the lumateperone patients with tremor, only 1 patient discontinued due to tremor that affected both hands, was mild in intensity, and resolved.
There were no TEAEs of mania or hypomania reported in either group.
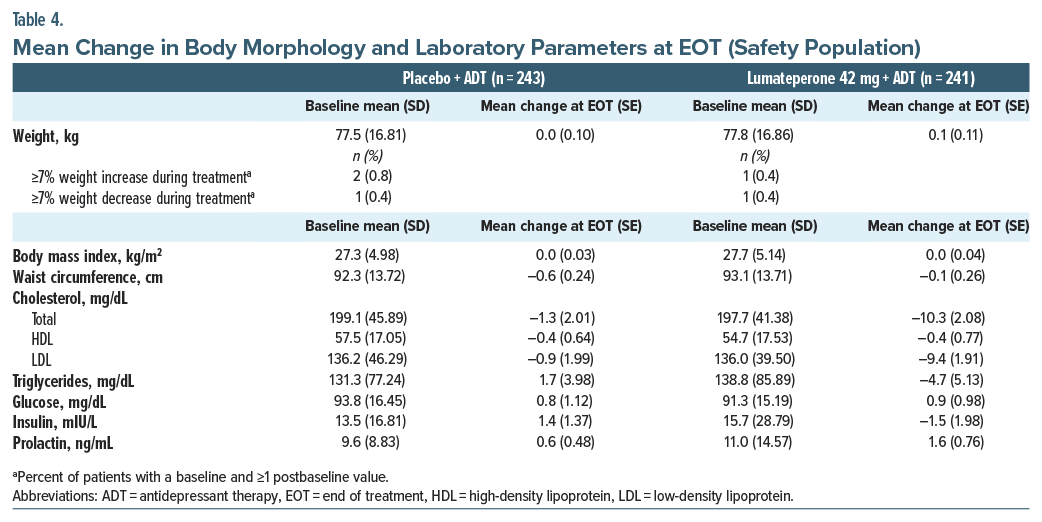
With lumateperone + ADT, there were no notable changes at the end of treatment in weight, body mass index, or waist circumference (Table 4). Changes in cardiometabolic parameters and prolactin were not clinically significant and were generally similar between groups (Table 4). No patients met criteria for Hy’s law. Potentially clinically significant weight increase or decrease (≥7% from baseline) was rare (<1%, Table 4).
In the placebo + ADT group, 1 patient each had a QT Fridericia-corrected interval ≥480 ms and an increase of >60 ms from baseline; no patients had an interval ≥500 ms. In the lumateperone + ADT group, no patients had QT Fridericia-corrected interval ≥480 ms or an increase of >60 ms from baseline.
According to the C-SSRS, no suicidal behavior occurred during treatment. Emergence of suicidal ideation was lower with lumateperone+ADT (n=3 [1.4%]) vs placebo+ADT (n=8 [3.5%]). No TEAEs of suicidal ideation occurred in the lumateperone+ADT group, vs 1 in the placebo+ADT group.
DISCUSSIONIn this phase 3, randomized, double-blind, placebo-controlled trial, lumateperone 42 mg+ADT significantly improved depression symptoms and disease severity vs placebo+ADT in patients with MDD and inadequate ADT response. Approximately 50% of patients with MDD experience inadequate ADT response, and lack of symptom management is associated with increased recurrence risk, more chronic depressive episodes, and worse functioning.32 Additionally, many therapies have a slow onset of action, requiring 6–12 weeks to achieve benefits which can further increase the illness burden.4 In this study, clinically meaningful, significant improvements in depression symptoms and disease severity occurred by day 8 and persisted throughout the study with lumateperone+ADT. At day 43, the MADRS Total score improvement with lumateperone+ADT from baseline compared with placebo+ADT (LSMD=–4.9) was of a slightly higher magnitude than that reported in other trials of antipsychotics in patients with MDD and inadequate response.33–41
Remission is an important goal of MDD treatment, as patients who fail to achieve remission have prolonged psychosocial impairment and higher relapse risk.2 With lumateperone 42 mg+ADT, MADRS response and remission rates at day 43 were significantly greater than those of placebo +ADT, with similar or greater fold improvements as those reported for approved adjunctive antipsychotics for MDD.33–37,40-42
Patient-reported outcomes provide valuable information about treatment effects from a patient’s perspective and ensure that the treatment is comprehensively assessed, capturing information that cannot be gathered from other outcomes.43 Thus, the inclusion of patient-reported outcomes in clinical trials has recently increased in frequency.43 Compared with placebo+ADT, at day 43 in this trial, lumateperone+ADT significantly improved in QIDS-SR-16 Total score, a patient-reported scale measuring depression severity, which strengthens the positive results of the clinician-rated end points in this study. Lumateperone+ADT also significantly improved the patient-reported anxiety scale GAD-7 at day 43 vs placebo+ADT. Anxiety commonly occurs in patients with MDD, and the presence of a comorbid anxiety disorder is a strong predictor of MDD severity.44 In addition to improving patient-reported anxiety, lumateperone+ADT significantly improved depression symptoms and severity vs placebo+ADT in the difficult-to-treat45 subgroup of patients with anxious distress.
The favorable safety of lumateperone+ADT in this trial is consistent with that reported in other trials of lumateperone in MDD with mixed features,46 bipolar depression,47,48 and schizophrenia,49–51 demonstrating minimal-to-low risk of EPS, weight gain, or cardiometabolic and prolactin abnormalities. Here, the majority of TEAEs were of mild-or-moderate severity, and the most common TEAEs were dry mouth, fatigue, and tremor. Dry mouth and fatigue are commonly reported in clinical trials with other approved antipsychotics adjunctive to ADT.35–37,42 In this study, all TEAEs of tremor were of mild-or-moderate severity and resolved or were resolving. No suicidal behavior occurred during lumateperone+ADT treatment, and emergence of suicidal ideation was low. There were no TEAEs of mania or hypomania indicating a low risk of emergent mania, which is of special importance for patients who have MDD with mixed features. Overall, these results highlight the favorable safety profile of lumateperone+ADT in patients with MDD and inadequate response.
A limitation of this study is the exclusion of patients with treatment-resistant illness, imminent suicidal risk, or comorbid psychiatric illnesses other than MDD, which may limit the generalizability of the findings. An additional limitation is the short-term duration; however, an open-label long-term study (NCT05061719) was completed in December 2024, and long-term data are being prepared for publication. In addition to the current trial, a similarly designed study (NCT05061706) also demonstrated clinically meaningful efficacy of lumateperone 42 mg+ ADT over placebo + ADT, with a favorable safety profile in patients with MDD and inadequate ADT response.52
Overall, lumateperone 42 mg adjunctive to ADT demonstrated significant, clinically meaningful efficacy over placebo adjunctive to ADT, improving depressive symptoms and disease severity as measured by clinician-rated and patient-reported outcomes in patients with MDD and inadequate ADT response. Lumateperone 42 mg+ ADT was generally well-tolerated, consistent with prior lumateperone trials, with minimal-to-low risk of EPS. Weight gain, cardiometabolic parameters, and prolactin levels were similar to placebo. These results suggest that lumateperone 42 mg adjunctive to ADT is a promising new treatment option for adults with MDD with inadequate ADT response.
Article InformationPublished Online: August 25, 2025. https://doi.org/10.4088/JCP.25m15848
© 2025 Physicians Postgraduate Press, Inc.
Submitted: February 20, 2025; accepted June 20, 2025.
To Cite: Durgam S, Earley WR, Kozauer SG, et al. Lumateperone as adjunctive therapy in patients with major depressive disorder: results from a randomized, double-blind, phase 3 trial. J Clin Psychiatry 2025;86(4):25m15848.
Author Affiliations: Intra-Cellular Therapies, a Johnson & Johnson Company, Bedminster, New Jersey (Durgam, Earley, Kozauer, Chen, Lakkis); Department of Psychiatry, University of Toronto, Toronto, Ontario, Canada (McIntyre); Department of Psychiatry, University of California, San Diego and Riverside; La Jolla, California (Stahl).
Corresponding Author: Suresh Durgam, MD, Intra-Cellular Therapies, a Johnson & Johnson Company, 135 US Highway 202/206, Ste 6, Bedminster, NJ 07921 ([email protected]).
Relevant Financial Relationships: Drs Durgam, Earley, Kozauer, Chen, and Lakkis are full-time employees of Intra-Cellular Therapies, a Johnson & Johnson company, and may hold equity in the company. Dr McIntyre has received research grant support from CIHR/GACD/National Natural Science Foundation of China (NSFC) and the Milken Institute and speaker/consultation fees from Lundbeck, Janssen, Alkermes, Neumora Therapeutics, Boehringer Ingelheim, Sage, Biogen, Mitsubishi Tanabe, Purdue, Pfizer, Otsuka, Takeda, Neurocrine, Neurawell, Sunovion, Bausch Health, Axsome, Novo Nordisk, Kris, Sanofi, Eisai, Intra-Cellular Therapies, a Johnson & Johnson company, NewBridge Pharmaceuticals, Viatris, AbbVie, and Atai Life Sciences. Dr Stahl has served as a consultant to Acadia, Alkermes, Allergan, AbbVie, Arbor Pharmaceuticals, Axovant, Axsome, Celgene, Concert, Clearview, EMD Serono, Eisai Pharmaceuticals, Ferring, Impel NeuroPharma, Intra-Cellular Therapies Inc, Ironshore Pharmaceuticals, Janssen, Karuna, Lilly, Lundbeck, Merck, Otsuka, Pfizer, Relmada, Sage Therapeutics, Servier, Shire, Sunovion, Takeda, Taliaz, Teva, Tonix, Tris Pharma, and Viforpharma and is a board member of Genomind; he has served on speakers bureaus for Acadia, Lundbeck, Otsuka, Perrigo, Servier, Sunovion, Takeda, Teva, and Vertex and has received research and/or grant support from Acadia, Avanir, Braeburn Pharmaceuticals, Eli Lilly, Intra-Cellular Therapies, a Johnson & Johnson company, Ironshore, ISSWSH, Neurocrine, Otsuka, Shire, Sunovion, and TMS NeuroHealth Centers.
Funding/Support: This study was funded by Intra-Cellular Therapies, a Johnson & Johnson company, Bedminster, NJ, USA.
Role of the Funders/Sponsors: Intra-Cellular Therapies, a Johnson & Johnson company, was responsible for the design, analysis, interpretation, and publication of this study.
Previous Presentation: Poster and oral presentation at the ECNP Annual Meeting; September 21–24, 2024; Milan, Italy. Encore poster presented at the Psych Congress Annual Meeting; October 29–November 2, 2024; Boston, Massachusetts. Encore poster presented at the NEI Annual Meeting; November 7–10, 2024; Colorado Springs, Colorado. Encore poster presented at the CNS Summit Annual Meeting; November 10–13, 2024; Boston, Massachusetts. Poster presented at the ACNP Annual Meeting; December 8–11, 2024; Phoenix, Arizona. Encore poster presented at ADAA Conference; April 3–5, 2025; Las Vegas, Nevada. Encore poster presented at AAPP Annual Meeting; April 27–30, 2025; Salt Lake City, Utah.
Data Availability: Data will be made available on reasonable request, subject to
review and meeting criteria.
Author Contributions: Drs Durgam, Earley, Kozauer, Chen, and Lakkis participated in
study design and study conduct. All authors participated in data analysis/
interpretation and writing/critical review.
Acknowledgments: The authors thank all study investigators, research staff, and
patients for their participation. Medical writing support was provided by Kendall
Foote, PhD, of Nucleus Global, an Inizio company, funded by Intra-Cellular Therapies,
a Johnson & Johnson company.
Supplementary Material: Available at Psychiatrist.com.
Comments (0)