
Remember me
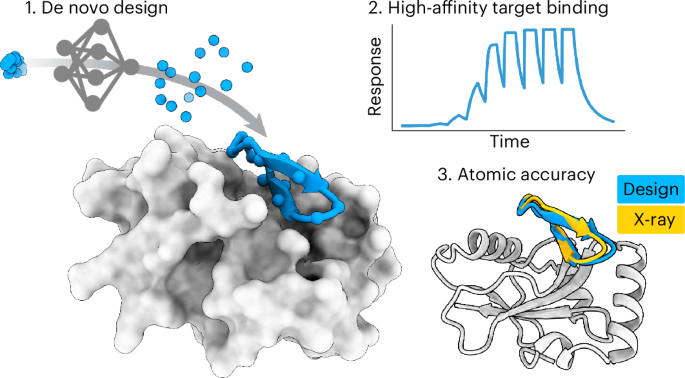

Macrocyclic peptide monomers and binders were designed with RFpeptides using a three-stage pipeline: backbone generation using RFdiffusion with the cyclic offset applied to the peptide chains, followed by sequence design using ProteinMPNN and, finally, structure prediction of the designed peptide–target complexes using either AfCycDesign and/or RoseTTAFold with the cyclic offset applied to the peptide. Designs were further filtered and downselected using Rosetta metrics and, in some cases, clustered on the basis of Cα r.m.s.d. Detailed computational methods, including example scripts, can be found in Supplementary Section 2.2.
Peptide synthesisMacrocyclic peptides described here were either purchased from Wuxi AppTec at greater than 90% purity or synthesized in-house using Fmoc-based solid-phase peptide synthesis. Peptides were typically synthesized on preloaded CTC resin. The resin was swollen in DCM followed by iterative deprotection with 20% piperidine in DMF and coupling with either HBTU (Sigma) or PyAOP (Novabiochem) and DIEA (Sigma). The linear peptides were cleaved from the resin using either 2% TFA in DCM or 20% HFIP (Oakwood Chemical) in DCM. The solvent was removed by rotary evaporation and linear protected peptides were cyclized in either DCM, DMF or a mixture of both depending on the solubility of the peptide, using two equivalents of PyAOP and five equivalents of DIEA overnight. The protecting groups were removed using a cocktail of 95:2.5:2.5, TFA, water and TIPS for 2.5 h. The crude peptides were precipitated using cold diethyl ether. The precipitate containing the crude cyclization reaction was dissolved in a mixture of water and acetonitrile for purification using reverse-phase high-performance liquid chromatography (LC). Peptide identities were confirmed by mass spectrometry (MS). Purities for all synthesized and tested macrocyclic peptides are also summarized in Supplementary Tables 7–10. The mass spectrograms and analytical LC chromatograms for all purified peptides are shown in Supplementary Section 4.
Protein expression and purificationMDM2 and MCL1The amino acid sequences of MCL1 (PDB 2PQK)37 and MDM2 (PDB 4HFZ)38 were retrieved from the PDB. The optimized genes were then cloned into a Novogen pRSF-DUET plasmid (Sigma, 71341-3), incorporating a 6xHis-tag at the N terminus, followed by an Avi-tag and a tobacco etch virus (TEV) protease cleavage site. The resulting constructs were codon-optimized for Escherichia coli expression and synthesized by Genscript. For propagation, the plasmids were transformed into E. coli NEBα cells (New England Biolabs, C2987); for protein expression, the plasmids were transformed into E. coli BL21(DE3) cells (New England Biolabs, C2527). A single sequence-verified colony was cultured in 50 ml of kanamycin (50 µg ml−1) selective Luria Broth (LB) medium. This culture was incubated at 37 °C with shaking at 200 rpm for 16 h overnight. Subsequently, 50 units of optical density at 600 nm (OD600) of the overnight culture were transferred to 1 L of fresh kanamycin (50 µg ml−1) selective LB medium. The culture was grown at 37 °C with shaking at 200 rpm for 2 h (until it reached an OD600 of 0.4–0.5), at which point the temperature was decreased to 20 °C. The culture was grown until an OD600 of 0.7–0.8; protein expression was induced by adding 1 mM IPTG and the culture was left to grow overnight for 14 h.
Cells were harvested by centrifugation at 5,000g for 10 min at 4 °C, resulting in a cell pellet with a density of 5 g L−1. The pellet was immediately flash-frozen and stored at −20 °C for later use. For lysis, the pellet was thawed on ice and resuspended in 5 ml of lysis buffer per gram of pellet. This lysis buffer contained 50 mM Tris-HCl, 300 mM NaCl and 10 mM imidazole and was supplemented with 1× BugBuster protein extraction reagent (Sigma-Aldrich, 70921), 200 µg ml−1 lysozyme (Sigma-Aldrich, L6876), 25 U per ml benzonase nuclease (Sigma-Aldrich, E8263) and 1× cOmplete EDTA-free protease inhibitor cocktail (Sigma-Aldrich, 11836170001). The buffer was filter-sterilized using a 0.2 µm filter before the addition of benzonase, mixed by inversion and kept on ice until use. Cells were completely resuspended in the lysis buffer using a homogenizer at low speed and incubated for 30 min at room temperature (22–25 °C). Following incubation, the suspension was sonicated using a Q500 Sonicator equipped with a four-tip probe. Sonication was conducted for 2–3 min using pulses of 10–15 s on followed by 10–15 s off at 70% amplitude. The lysate was clarified by centrifugation at 16,000g for 20 min.
Ni-NTA agarose resin (Qiagen, 30210) was equilibrated with 20 column volumes (CV) of ultrapure water, followed by 20 CV of equilibration buffer (50 mM Tris-HCl, 300 mM NaCl and 10 mM imidazole). Then, 4 ml of 50% resin suspended in equilibration buffer was used to bind His-tagged proteins from 25 ml of clarified lysate. All immobilized metal affinity chromatography (IMAC) steps were conducted at 4 °C. The lysate–resin mixture was incubated for 60 min on a rotary shaker set to a slow speed. After incubation, the resin was transferred to a 20-ml gravity column and allowed to completely settle. The resin was first washed with 20 CV of wash buffer 1 (20 mM Tris-HCl, 250 mM NaCl, 10 mM imidazole and 5 mM β-mercaptoethanol), followed by another 20 CV of wash buffer 2 (20 mM Tris-HCl, 500 mM NaCl and 35 mM imidazole). The bound proteins were then eluted with 8 ml of elution buffer (20 mM Tris-HCl, 250 mM NaCl, 350 mM imidazole and 2 mM DTT). Aliquots of the eluate were collected and analyzed using SDS–PAGE gels.
The eluate was loaded onto a pre-equilibrated Superdex 75 10/300 GL column (25 mM Tris-HCl, 250 mM NaCl and 2 mM DTT) and run at a flow rate of 0.6 ml min−1 using an ÄKTA pure system for size-exclusion chromatography (SEC). Then, 1 ml fractions were collected from the elution volume of 8–16 ml and those corresponding to peaks in the absorbance at 280 nm between an elution volume of 10 and 13 ml were assessed with SDS–PAGE gels. Fractions confirming the expected molecular weight were pooled and concentrated by centrifugation at 4,000g for 30 min at 4 °C using Amicon Ultra-4 concentrators with a 3 kDa cutoff (Millipore Sigma, UFC800308) to a final volume of 500 µl. The identity of the eluted proteins were confirmed by MS using an Agilent 6230 LC–MS time-of-flight system.
Verified protein samples were processed for further applications: biotinylation for SPR analysis or tag removal by TEV protease cleavage for crystallography. Biotinylation was performed using the BirA biotin–protein ligase standard reaction kit (Avidity, BirA-500) according to the manufacturer’s recommended conditions. The reaction was carried out at 4 °C overnight on a slowly shaking platform. For TEV protease cleavage, the proteins were treated with a 25:1 protein to TEVd enzyme ratio39. Similarly, the mixture was incubated at 4 °C overnight on a slowly shaking platform. Following these treatments, samples underwent a cleanup step using 1 ml of Ni-NTA resin per 20 mg of protein. The resin was pre-equilibrated with 10 CV of ultrapure water and 10 CV of a buffer containing 25 mM Tris-HCl, 250 mM NaCl and 10 mM imidazole. The pre-equilibrated resin was added to the protein mixture and incubated for 30 min on a rolling platform at 4 °C. Subsequently, the mixtures were filtered through a 0.45 µm PVDF centrifugal filtering unit to remove the Ni-NTA-bound substrates. The eluate was collected and dialyzed in 2 L of 25 mM Tris-HCl, 250 mM NaCl and 2 mM DTT using a Slide-A-Lyzer G3 dialysis cassettes with a 3.5 kDa molecular weight cutoff (Thermo Scientific, A52966) overnight for 18 h at 4 °C stirring. The dialyzed protein was concentrated to 0.2–0.5 ml (as required for downstream assays), using the Amicon ultra concentrators (as above), aliquoted and flash-frozen. Fractions were analyzed by mass spectroscopy for the efficacy of the biotinylation and TEV protease cleavage treatments, as previously described.
GABARAP for SPRA synthetic complementary DNA was designed on the basis of the amino acid sequence of GABARAP (UniProt O95166) and optimized for expression in E. coli using Benchling software. The construct was devised to include an N-terminal Avi-tag and TEV protease cleavage site and was cloned into the Novogen pET-50b(+) plasmid. This plasmid configuration introduced a tandem arrangement of protein tags at the N terminus: a 6xHis-tag, followed by a NusA solubility tag, another 6xHis-tag and a human rhinovirus (HRV) 3C protease cleavage site. Therefore, the final construct sequence was as follows: 6xHis–NusA–6xHis–HRV 3C–Avi–TEV–GABARAP. NusA was specifically chosen as a solubility tag because of its known effectiveness in enhancing protein solubility in E. coli40,41. The construct was synthesized and cloned by Genscript.
As described above for MCL1 and MDM2 protein expression, the plasmids were introduced into E. coli NEBα cells and BL21(DE3) cells. A single sequence-verified colony was cultured in 50 ml of kanamycin (50 µg ml−1) selective LB medium for 16 h at 37 °C, shaking at 200 rpm. Then, 50 OD600 units of this culture were transferred to 1 L of fresh kanamycin (100 µg ml−1) selective autoinduction medium (TBM-5052: 1.2% (w/v) tryptone, 2.4% (w/v) yeast extract, 0.5% (v/v) glycerol, 0.05% (w/v) d-glucose, 0.2% (w/v) d-lactose, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 10 μM FeCl3, 4 μM CaCl2, 2 μM MnCl2, 2 μM ZnSO4, 400 nM CoCl2, 400 nM NiCl2, 400 nM CuCl2, 400 nM Na2MoO4, 400 nM Na2SeO3 and 400 nM H3BO3). The culture was grown at 37 °C with shaking at 200 rpm for 2 h, at which point the temperature was decreased to 22 °C and the culture was left to grow for 16 h.
Cells were harvested, lysed and purified following the protocol outlined earlier for MCL1 and MDM2, with some modifications. The cultures yielded a cell pellet amounting to 15 g L−1. Lysis was completed using an IKA T18 microfluidizer at 450 psi, followed by lysate clarification by centrifugation at 16,000g for 15 min. All IMAC steps were conducted at 22 °C, except for the incubation of the lysate–resin mixture, which was performed at 4 °C. Proteins bound to the resin were eluted with 5 ml of elution buffer (50 mM Tris-HCl pH 8, 250 mM NaCl and 300 mM imidazole). SEC was then performed using a Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated with TBS (50 mM Tris-HCl pH 8 and 250 mM NaCl). Fractions confirmed by SDS–PAGE were pooled and concentrated using Amicon Ultra-15 concentrators with a 30 kDa cutoff (Millipore Sigma, UFC9030) to a final volume of 1 ml. Downstream processing for SPR analysis was performed as described previously, with one modification. For biotinylation, the protein was first cleaved using HRV 3C protease with the reagents and protocol provided by the Pierce HRV 3C protease solution kit (Thermo Scientific, 88946). The digested samples were subsequently purified and verified, as outlined in earlier sections.
GABARAP and GABARAPL1 for crystallographyGABARAP and GABARAPL1 were expressed as glutathione S-transferase (GST) fusion proteins after transforming E. coli BL21(DE3) T1 cells with pGEX4T2-GABARAP and pGEX4T2-GABARAPL1 plasmids, respectively. Bacteria were cultivated in LB medium containing 100 µg ml−1 ampicillin; gene expression was induced with 1 mM IPTG at an OD600 of 0.6–0.8 and allowed to proceed for 20 h at 25 °C. Afterward, cells were harvested by centrifugation at 3,000g for 30 min at 4 °C. The bacterial pellet was washed with PBS (137 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4 and 10 mM Na2HPO4) and resuspended in lysis buffer (PBS supplemented with 5% (v/v) glycerol, 0.01% (v/v) β-mercaptoethanol, 10 µg ml−1 DNase (AppliChem, A3778) and cOmplete EDTA-free protease inhibitor cocktail (Roche, 11836170001)) before application to the cell disruptor (Constant Systems, model TS1.1) for three cycles with 1.9 kbar at 4 °C. Lysates were cleared by centrifugation at 4 °C with 45,000g for 45 min. The GST fusion proteins were purified from the supernatant by affinity chromatography using glutathione Sepharose 4B (Cytiva, 1705605). Cleavage with thrombin (Sigma-Aldrich, 1.12374) during dialysis against 10 mM Tris-HCl and 150 mM NaCl (pH 7.0) at 4 °C overnight yielded 119 amino acid proteins carrying an N-terminal Gly-Ser extension in addition to the native residues of GABARAP and GABARAPL1. Subsequently, samples were applied to a Hiload 26/60 Superdex 75 preparatory-grade size-exclusion column (GE Healthcare) equilibrated with 10 mM Tris-HCl and 150 mM NaCl (pH 7.0). Protein purity was assessed by SDS–PAGE and Coomassie staining. Fractions containing the eluted proteins were concentrated to 3–5 mg ml−1 using Vivaspin 20 concentrators with a 3 kDa cutoff (Sartorius), flash-frozen in liquid N2 and kept at −80 °C for long-term storage.
RbtA β-helix domainFor heterologous expression of the β-helix domain of RbtA (residues A20–I459) in E. coli, the gene was amplified and fused with a SNAC tag (GSHHWGS) at the C terminus using the following primers: forward, GCTGCCCAGCCGGCGATGGCCATGGGCGCTGATATTGAAGTCACAACTAC; reverse, CAGTGGTGGTGGTGGTGGTGCTCGAGGCTGCCCCAATGATGGCTGCCGATATATTCAATTGCGCCTAAAT42. The fragment was inserted into NcoI-digested and XhoI-digested pET-22b(+) by Gibson assembly to generate a construct with a C-terminal 6xHis fusion. The construct was confirmed by sequencing and transformed into E. coli Rosetta (DE3) cells.
To purify the β-helix domain of RbtA, an overnight culture of Rosetta (DE3) cells carrying the construct was back-diluted 1:300 in 2× YT broth and grown at 37 °C with shaking at 200 rpm until the OD600 reached 0.4. The incubation temperature was reduced to 18 °C, IPTG was added to a final concentration of 0.3 mM and the culture was incubated for a total of 18 h. Cells were then collected by centrifugation and resuspended in lysis buffer containing 200 mM NaCl, 50 mM Tris-HCl pH 7.5, 10% glycerol (v/v), 5 mM imidazole, 0.5 mg ml−1 lysozyme and 1 mU of benzonase. Cells were then lysed by sonication and cellular debris was removed by centrifugation at 35,000g for 30 min at 4 °C. The protein was purified from lysates using a 1 ml HisTrap HP column on an ÄKTA fast protein LC (FPLC) system. Column-bound protein was eluted using a linear imidazole gradient from 5 to 500 mM. Protein purity was assessed by SDS–PAGE and Coomassie staining. The fractions with high purity were concentrated using a 30 kDa cutoff Amicon filter and then further purified by FPLC using a HiLoad 16/600 Superdex 200 preparatory-grade column (GE Healthcare) equilibrated with sizing buffer (500 mM NaCl, 50 mM Tris-HCl pH 7.5 and 10% glycerol (v/v)). The fractions with high purity were concentrated and used for evaluation of macrocyclic binders or determination of X-ray structure.
For determination of the X-ray crystal structure of RbtA, the C-terminal 6xHis-tag was removed by chemical cleavage at the SNAC tag. In brief, the buffer of the concentrated protein was exchanged to cleavage buffer (0.1 M CHES, 0.1 M NaCl, 0.1 M acetone oxime and 5 mM Fos-choline-12, pH 8.6). The protein solution was diluted to 1 mg ml−1, followed by the addition of 1 mM TCEP and 1 mM NiCl2. The mixture was vortexed and incubated at room temperature for 16 h. The precipitation was removed by centrifugation at 35,000g for 30 min at 4 °C. The supernatant was concentrated and exchanged to Tris buffer (50 mM Tris-HCl pH 7.5 and 200 mM NaCl). The protein solution was incubated with a 1 ml bed volume of Ni-NTA beads to extract the cleaved 6xHis-tag. The resulting fraction was concentrated and then further purified by FPLC using a HiLoad 16/600 Superdex 200 preparatory-grade column.
Crystallization of protein–cyclic peptide complexesMCL1 with cyclic peptideMCL1 (18.5 mg ml−1) and macrocycle MCB_D2 were mixed in 1:2 molar ratio and incubated for 30 min at room temperature. Upon addition of the MCB_D2 to the protein, we observed some precipitation. This precipitant was removed by centrifugation before crystallographic screening. Crystallization experiments for the MCL1–MCB_D2 complex were conducted using the sitting-drop vapor diffusion method. Initial crystallization trials were set up in 200 nl drops using 96-well crystallization plates. Crystal drops were imaged using the UVEX crystal plate hotel system by JANSi. Diffraction-quality crystals for the complex appeared in 0.2 M sodium chloride, 0.1 M Bis–Tris pH 6.5 and 25% (w/v) polyethylene glycol 3350 (Hampton Research) in 2 weeks.
GABARAP and GABARAPL1 with cyclic peptidesCyclic peptides GAB_D8 and GAB_D23 were dissolved in 10 mM Tris-HCl and 150 mM NaCl (pH 7.0) and each mixed with both GABARAP and GABARAPL1, targeting a peptide-to-protein molar ratio of 3:2. After incubation for 10 min at room temperature, any insoluble components were removed by centrifugation (10 min at 20,000g and 4 °C). The protein–peptide complexes were concentrated using Amicon Ultra-0.5 centrifugal filter units with a 3 kDa cutoff (Merck) until a final protein concentration of 6–8 mg ml−1 (GABARAPL1–GAB_D8) or 13–15 mg ml−1 (GABARAP–GAB_D23) was reached. Samples were once again cleared of particles by centrifugation (30 min at 20,000g and 4 °C) before application in crystallization experiments. Search for crystallization conditions was performed by the sitting-drop vapor diffusion method using robotic systems Freedom Evo (Tecan) and Mosquito LCP (SPT Labtech) with commercially available screening sets. Experiments were set up by combining 200 nl of protein–peptide complex with 100 nl (for GABARAPL1–GAB_D8) or 200 nl (for GABARAP–GAB_D23) of reservoir solution and plates were incubated at 20 °C. Crystals appeared for a number of conditions, which were subjected to optimization as appropriate. Diffraction-quality samples used for X-ray structure determination developed with reservoir solutions containing 0.17 M ammonium sulfate, 25.5% (w/v) PEG 4000 and 15% (v/v) glycerol for GABARAPL1–GAB_D8 and 0.1 M MES pH 5.0 and 30% (w/v) PEG 6000 in the case of GABARAP–GAB_D23. Diffraction data (https://doi.esrf.fr/10.15151/ESRF-DC-1966164200 and https://doi.esrf.fr/10.15151/ESRF-DC-1979522808 for GABARAPL1–GAB_D8 and GABARAP–GAB_D23, respectively) were collected at 100 K on beamline BM07 of the European Synchrotron Radiation Facility (ESRF) tuned to an X-ray wavelength of 0.9795 Å, using a Pilatus 6M detector (DECTRIS). Data processing was carried out with XDS and XSCALE43 and included reflections up to a diffraction limit of 1.5 Å for GABARAP–GAB_D23 and 2.5 Å for GABARAPL1–GAB_D8. The GABARAP–GAB_D23 structure featuring space group C2 was determined by molecular replacement (MR) using MOLREP44 with the structure of GABARAP from its K1 peptide complex (PDB 3D32)30 as a template. For the GABARAPL1–GAB_D8 complex, initial evaluation suggested tetragonal symmetry but with strong indications of twinning. Data integration in maximal translationengleiche subgroups followed by MR search using MoRDa45 revealed P212121 as the true space group, with near-perfect pseudomerohedral twinning accounting for apparent Laue group 4/mmm. To avoid bias in cross-validation, this pseudosymmetry of the data was explicitly accounted for in flag assignment. The solution obtained for GABARAPL1–GAB_D8 was subjected to a round of automated rebuilding in phenix.autobuild46. In either case, model refinement was performed with phenix.refine47, alternating with interactive rebuilding in Coot48, which included stepwise introduction of cyclic peptides GAB_D8 and GAB_D23. According to validation using MolProbity49 and the wwPDB validation system (https://validate-rcsb-2.wwpdb.org/), both models featured good geometry. Detailed statistics of data collection and refinement can be found in Supplementary Table 6.
RbtA with cyclic peptide and apo RbtARbtA (10 mg ml−1) and RBB_D10 were mixed in a 1:5 molar ratio and incubated for 30 min at room temperature. Initial crystallization trials were set up in 200 nl drops using 96-well crystallization plates and the experiments were conducted by the sitting-drop vapor diffusion method. Crystal drops were imaged using the UVEX crystal plate hotel system by JANSi. Diffraction-quality crystals for the RbtA–RBB_D10 complex appeared in 0.2 M lithium sulfate, 0.1 M Tris pH 8.5 and 40% (v/v) PEG 400 (JCSG Plus, Hampton Research). Additionally, we soaked the crystals in 22.32 mg ml−1 RBB_D10 for 5 min before flash-freezing. Crystals for RbtA alone (18.7 mg ml−1) were grown in 0.1 M Bis–Tris pH 6.5 and 20% (v/v) PEG 5,000 MME (SG1, Molecular Dimensions). All crystals were flash-cooled in liquid nitrogen before shipping to the synchrotron for data collection.
Diffraction data were collected at the NSLS2 beamline AMX/FMX (17-ID-1/17-ID-2). X-ray intensities and data reduction were evaluated and integrated by XDS43 and merged and scaled by Pointless and Aimless in the CCP4i2 program suite50. The X-ray crystal structure was determined by MR using the designed model for phasing by Phaser51. Next, the structure obtained from the MR was improved and refined by Phenix47. Model building was performed by Coot48 in between the refinement cycles. The final model was evaluated by MolProbity49. Data collection and refinement statistics are reported in Supplementary Table 5.
SPRSPR experiments were performed using a Cytiva Biacore 8K in HBS-EP+ buffer from Cytiva. Measurements were obtained by immobilization of biotinylated target protein using the biotin capture kit from Cytiva. Binding screens were performed by single-cycle kinetics experiments using the standard protocol in the Biacore 8K control software at 30 µl min−1 with serial injections of 10 nM, 100 nM, 1 µM, 10 µM and 100 µM, an association time of 60 s and a dissociation time of 120 s. For MCL1 designs, a dissociation time of 150 s was used. To evaluate the affinity of successful designs, a nine-point single-cycle kinetics experiment was performed with an association time of 90 s and dissociation time of 300 s. The dilution series for MCB_D2 was twofold starting at 20 µM, that for MDB_D8 was fivefold starting at 50 µM, and those for GAB_D8, GAB_D23 and RBB_D10 were fivefold starting at 20 µM. Reported measurements were analyzed using Biacore Insight evaluation software; sensorgrams were double-referenced and fit with a 1:1 binding kinetics fit model.
AlphaScreen assayWe used the AlphaScreen assay as described by Leveille et al.52 to measure inhibition of the GABARAP–K1 interaction by the computationally designed macrocycles. K1 is a previously described GABARAP binder with a Kd of 10 nM (ref. 27). Biotin-labeled peptide K1 was used at a final concentration of 10 nM and incubated with 10 nM (final concentration) of 6xHis–GABARAP in a final reaction volume of 50 μl. Computationally designed inhibitor peptides were serially diluted with 1:3 dilutions using the highest final concentration of 50 μM and added to the reaction mixture. The buffer used was 25 mM HEPES pH 7.3, 150 mM NaCl, 0.01% Tween, 1 mg ml−1 BSA and 0.5% DMSO. The plate was covered in foil, centrifuged at 1,500 rpm for 2 min and incubated for 150 min at room temperature with shaking. Then, 20 μg ml−1 (final concentration) of the streptavidin donor beads and nickel chelate acceptor beads were added in the dark before incubating for another 45 min. Data were collected on a Tecan plate reader using excitation at 680 nm and emission at 520–620 nm. Data were normalized to 0% (buffer only) and 100% (protein and tracer peptide, no inhibitor) controls. IC50 values were obtained from curve fits using GraphPad Prism 9 software, using the equation \(Y=\frac}_})}^)}\), where X is the concentration of inhibitor and h is the Hill coefficient. At least three independent replicates were used to calculate the average IC50 and the s.e.m.
Statistics and reproducibilityNo statistical method was used to predetermine sample size. One trial from the AlphaScreen that was used to determine the IC50 of GAB_D8 was repeated and the repeated value is what was used. All data are included in the Source Data. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)