
Remember me


Dilated cardiomyopathy (DCM) is a condition in which adverse eccentric remodeling due to non-ischemic disorders results in impaired contractile function. There are currently 19 genes associated with DCM [16, 17]. The location of these gene products is diverse and includes the sarcomere, junctional membrane, Z disc, sarcoplasmic reticulum, desmosome, cytoskeleton, nuclear envelope, ion channel, and RNA binding [3, 17]. A four-year multicenter study found that 32% of genotype-positive DCM patients experienced major events, 16% developed end-stage heart failure, and 20% had malignant arrhythmias [18].
CMR can provide insights into cardiac function, morphology, and tissue characteristics to diagnose and monitor dilated cardiomyopathy. Elevated end-diastolic diameter > 117% of the predicted value adjusted for age and body surface area or dilated ventricular volumes are hallmarks of DCM. CMR can detect subclinical myocardial dysfunction through global ventricular and atrial strain, which is often abnormal in DCM. Myocardial crypts, papillary muscle anomalies, and muscular bands may indicate genetic variants, regional wall motion abnormalities due to fibrosis, and excessive trabeculations secondary to remodeling can be seen in DCM, particularly those involving sarcomeric mutations [19].
In the early stages of DCM, interstitial fibrosis can develop from adverse remodeling, which can be detected based on the elevation of native T1 and ECV. The stepwise increases in T1 values correlate with a higher risk of adverse outcomes [20]. The progression of fibrotic changes can manifest in DCM as linear mid-wall septal LGE, which is associated with an increased risk of all-cause mortality, sudden cardiac death, cardiovascular hospitalization, and ventricular tachycardia in DCM patients [21]. In addition, patients can have a ring-like pattern or transmural involvement of LGE, often linked to DSP, FLNC, and PLN mutations, and have a worse prognosis with a higher risk of arrhythmia [22].
In a meta-analysis of 19 studies (7,330 DCM patients), the authors found LGE to be a better outcome predictor than ejection fraction alone. A 1% increase in LGE extent was associated with a 10% increase in all-cause mortality. LGE location (mid-wall with/without free wall) and pattern (mid-wall, sub-epicardial, and focal) were linked to worse outcomes. The presence of LGE was associated with a higher arrhythmia risk in patients with LVEF ≥ 35% (HR: 5.79) compared to those with LVEF < 35% (HR: 4.49) [12, 23].
There have been imaging features and specific gene-associated DCM subtypes [19]. One subtype of interest is LMNA cardiomyopathy. CMR identifies interstitial fibrosis as an early marker, preceding clinical symptoms and LGE which are seen in advanced disease. Mid-wall LGE indicates a poor prognosis. Elevated ECV is found in LMNA mutation carriers with normal LV function and in some without LGE. Other gene-specific LGE and CMR findings are shown in Table 1.
Table 1 Types of genetic cardiomyopathies with known associated genetic mutations/alleles, cellular locations or associated proteins, patterns of late gadolinium enhancement (LGE), and advanced cardiac magnetic resonance (CMR) features, including parametric mapping (T1, T2, T2* and extracellular volume (ECV) fraction values)DCM can resemble myocarditis with similar LGE patterns and elevations in T2. Pathogenic genetic variations can be seen in about 22% of adults and 44% of children with myocarditis and concomitant heart failure or ventricular arrhythmias [24]. This observation is thought to arise from the interaction between genetic predispositions and environmental triggers like infections [25]. For example, TTN truncating variants may impair the stress response, contributing to the transition from inflammation to full-blown DCM, while mutations in the DSP gene, common in arrhythmogenic cardiomyopathy, may predispose the heart to inflammation due to defects in cardiac desmosomes. In these cases, myocardial inflammation could act as a"second hit,"triggering or exacerbating the phenotypic expression of genetic cardiomyopathies [24]. These CMR and genetic findings suggest a possible inflammatory pathway leading to DCM and a potential therapeutic target that requires further exploration.
Hypertrophic CardiomyopathyHypertrophic cardiomyopathy (HCM) is the most common inherited cardiomyopathy with a prevalence of 1:200–1:500. HCM is characterized by maximal left ventricular (LV) wall thickness ≥ 15 mm without another cause, or ≥ 13 mm with a family history or genotype. Thought of as a genetic disease of the sarcomere, over 1400 mutations have been identified in at least 8 causative genes. The most common genes are myosin heavy chain 7 (MHY7) and myosin-binding protein C (MYBC3). Despite this, only ~ 40% of patients have an identifiable genetic mutation. Another 20–30% exhibit autosomal dominant inheritance without a clear causative gene [26].
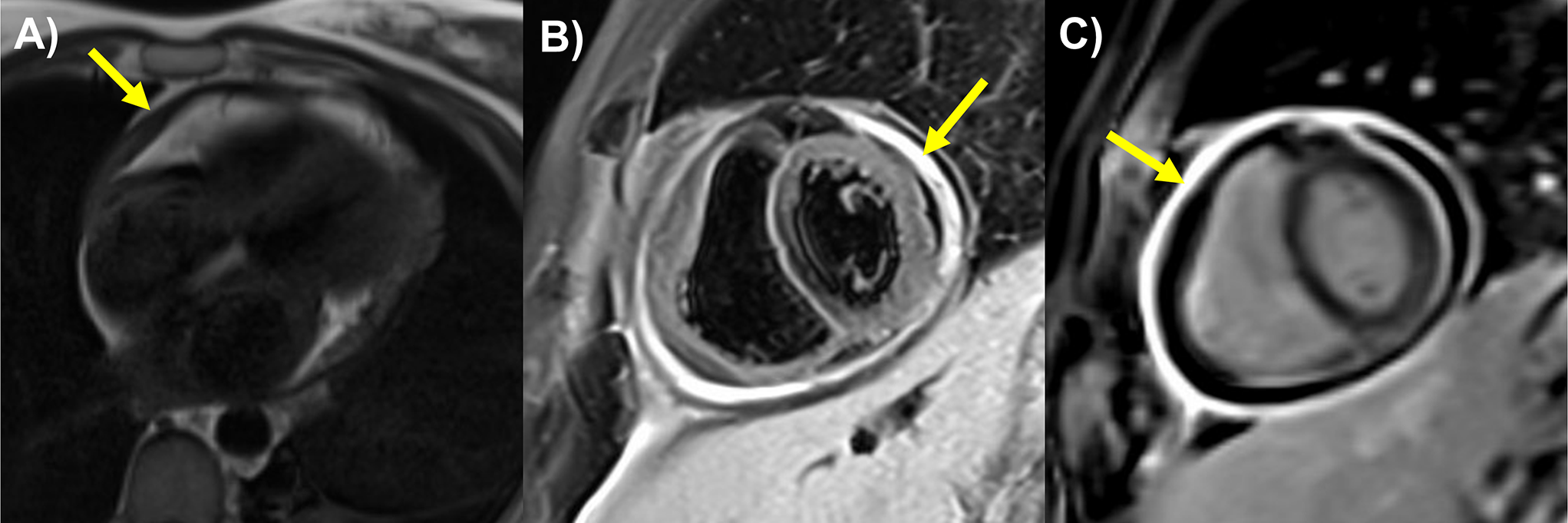
According to the Hypertrophic Cardiomyopathy Registry, there are two main subtypes of HCM. The first is the isolated basal septal hypertrophy subtype, which accounts for 40–50% of cases. In this group, pathogenic mutations are rare (~ 10%) and patients are typically older. In addition, they have more left ventricular outflow tract obstruction but lower sudden cardiac death (SCD) risk. The second is the reverse curvature hypertrophy subtype. This group is characterized by mid-septal thickening and a counter-clockwise “spiraling” hypertrophy pattern as the myocytes traverse from the LV base to the apex as seen in the short axis plane of the ventricle. Hypertrophy starts at the basal anterior wall, increasing to maximal thickness in the mid-septum, and then continuing into the mid-inferior segments (Fig. 1). Up to 90% of this subtype has an identifiable genetic mutation. There is a lower incidence of left ventricular outflow tract obstruction but a higher SCD risk [27]. A rarer subtype is apical HCM, which accounts for 5–10% of cases in the United States. They can have a favorable prognosis, but the development of mid-cavity obstruction and aneurysm drastically increases the risk of thrombus and arrhythmias [28, 29]. Additional HCM subtypes include those with concentric hypertrophy, neutral septal pattern, right ventricle (RV) predominance, and isolated lateral wall involvement [29].
Fig. 1
Hypertrophic cardiomyopathy. Reverse curvature phenotype hypertrophic cardiomyopathy stress CMR: a) Systolic anterior motion of the anterior mitral valve leaflet (SAM) observed on SSFP cine image on 4 CH (4 Chamber) view (yellow arrow). b) maximum wall thickness of 24 mm in the mid-short axis (SAX) cine image in the end of diastole (yellow arrow). c) short axis phase-sensitive inversion recovery (PSIR) late gadolinium enhancement (LGE) image: dense patchy LGE observed in the mid inferoseptum (yellow arrow). d) long axis PSIR LGE image: patchy LGE in the mid inferoseptum on 4 CH view (yellow arrow). e) native T1 mapping showed increased T1 relaxation time (1124 ms) in the mid inferoseptum (white arrow). f) Extracellular volume (ECV) image showed increased ECV value (39%) in mid inferoseptum (white arrow). g) rest perfusion pixel-wise mapping image with global resting myocardial blood flow (MBF) of 1.24 ml/g/min. h) stress MBF pixel-wise mapping image with low stress MBF (0.81 ml/g/min) in the mid septum (white arrow) and normal stress MBF (2.52 ml/g/min) in the mid lateral walls (green arrow), Myocardial perfusion reserve (MPR) in mid septum: 0.85, MPR in mid lateral walls: 2.31
According to the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC) 2023 guidelines, CMR has a class I recommendation for evaluating HCM. Specifically, CMR can be used to confirm myocardial thickness, quantify LGE burden, assess ejection fraction, help with procedural planning, and rule out alternative diagnoses [17, 30]. CMR combined with genetic testing can differentiate HCM from mimics like hypertensive or athletic remodeling, amyloidosis, and other cardiomyopathies. For patients with borderline maximal LV thickness that do not meet the criteria of HCM, additional CMR features may provide supporting evidence for a sarcomere disorder. Findings such as elongation of the anterior leaflet, myocardial clefts, and abnormal papillary muscles may be antecedent features in HCM [31]. Regarding specific genes, ALKP3 truncating variants can often be identified based on their distinct phenotypes. Heterozygotes show mixed concentric and apical hypertrophy with extensive LGE, while homozygotes develop early-onset dilated cardiomyopathy and later HCM-like hypertrophy [32]. However, CMR cannot distinguish between the more common genotypes such as MYH7 and MYBPC3.
Data on genotype and outcomes are mixed, but CMR LGE imaging of scars is an important prognostication tool. Most agree that sarcomere mutation-positive patients exhibit more fibrosis on CMR [33]. Ninety percent of HCM cases involve the septum, especially the commonly thickened segments such as the basal anterior or anteroseptal regions (Fig. 1). When quantifying replacement fibrosis in HCM with CMR [25, 26], LGE greater than 10–15% of the myocardial mass is associated with a higher mortality risk. CMR can also detect ventricular aneurysms and assess systolic function, which can help guide decisions regarding implantable cardioverter-defibrillator (ICD) placement according to ACC/AHA guidelines [30]. Deep learning has also been applied to CMR and HCM to identify scars without the need for GBCAs, which is known as virtual native enhancement imaging. This technique could help reduce cost and scan time while improving accessibility for HCM patients [34]. Quantitative CMR perfusion imaging has also been studied in HCM, demonstrating microvascular dysfunction based on reduced MBF and MPR (Fig. 1). Interestingly, these abnormalities are observed even in regions without hypertrophy or fibrosis [35].
Arrhythmogenic CardiomyopathyArrhythmogenic cardiomyopathy (ACM) is a genetic myocardial disease characterized by fibro-fatty replacement of myocardium and associated ventricular arrhythmias and sudden cardiac death. CMR is the imaging modality of choice due to its ability to provide detailed morpho-functional and tissue characterization of both ventricles. There are multiple criteria for diagnosing ACM. The 2010 Task Force criteria are specific for AMC with primarily RV involvement, while the 2020 Padua criteria also include left-dominant phenotypes with additional CMR structural abnormalities [36, 37]. The European Task Force consensus report published in 2024 proposed incorporating more LGE-based CMR findings, increased recognition of left-sided and biventricular phenotypes, and routine integration of genetic findings [22, 36].
There are many CMR features to evaluate for individuals with suspected ACM. RV-predominant phenotypes can have LGE primarily in basal RV regions (lateral/inferior walls), sparing the apex. In the RV, wall motion abnormalities like akinesia, dyskinesia, or aneurysms may occur. However, the relatively thin wall of the RV compared to the LV can decrease the sensitivity of these findings. Therefore, combining LGE information with motion abnormalities is recommended to improve diagnostic accuracy. In LV-predominant ACM, LGE typically appears as subepicardial or mid-myocardial scarring (Fig. 2), especially in the inferolateral wall. Both LV and RV ACM can have regional hypokinesis despite normal ventricular volumes. Biventricular ACM combines both features RV and LV phenotypes, with LGE and segmental wall motion abnormalities extending to both chambers. Aquaro et al. demonstrated CMR detected ACM with biventricular involvement is associated with a worse prognosis, with an increased risk of fatal arrhythmias compared to RV involvement alone. However, patients diagnosed with ACM based on the revised Task Force criteria but no evidence of the disease on CMR had a favorable prognosis with no major events over 5-years [40].
Fig. 2
Arrhythmogenic cardiomyopathy with DSP pathogenic gene: a) short axis (SAX) cine image, b) short axis late gadolinium enhancement (LGE) image: LGE stripe in the mid septum (yellow arrow). c) ECV (extracellular volume) mapping showed diffusely increased ECV (32%), d) long axis (LAX) 3 chamber (3 CH) cine showed severely enlarged left ventricular size and LVEF 26%. e) long axis phase-sensitive inversion recovery (PSIR) LGE image: mid wall stripe of LGE in septum on 3 CH view (yellow arrows). f) T2 image showed increased T2 value (56 ms) (green arrows)
The different subtypes of ACM are associated with distinct genetic variants. RV involvement is seen in genetic mutations such as Plakophilin- 2 (PKP2), Desmocollin- 2 (DSC2), and Junction Plakoglobin (JUP). LV predominance is associated with Desmoplakin (DSP) (Fig. 2), Lamin (LMNA), Filamin C (FLNC), and Transmembrane Protein 43 (TMEM43). Finally, biventricular involvement occurs with genes such as Desmoglein- 2 (DSG2), Desmin (DES), and Phospholamban (PLN) [38]. Variants in the TMEM43 gene were associated with a high prevalence of subepicardial late gadolinium enhancement and left ventricular dysfunction on CMR images [39].
Research has shed light on the role of inflammation and immunopathology in ACM and other genetic cardiomyopathies. Histological studies, reported by Thiene and Basso, reveal inflammatory infiltrates of lymphocytes surrounding necrotic myocytes in two-thirds of ACM hearts, indicating immune activation. It is postulated that autoimmune mechanisms may further amplify disease progression. Validated across studies, increased DSG2 autoantibodies have been shown to result in impaired gap junction function and increased arrhythmia burden [40]. In ACM, TGFβ3 signaling is an important driver of fibrotic remodeling, while NFκB and GSK3β pathways exacerbate inflammatory responses and desmosomal instability [41]. A multicenter study by Corrado et al. found that PKP2 dysregulation is a key link between cytokine release and structural destabilization [42]. A study by Peretto et al. showed that immunomodulatory therapy can potentially be used to treat inflammation in cardiomyopathies linked to the DSP (Fig. 2), TTN, FLNC, PKP2, LMNA, and SCN5 A genes. Based on imaging and biopsy, they demonstrated that immunomodulatory therapy cleared inflammation in 67% of subjects, potentially reversing genetic inflammatory pathways [43].
Inherited Cardiac AmyloidosisHereditary transthyretin amyloidosis (ATTR) is caused by an autosomal-dominant transthyretin gene variant, which leads to progressive misfolded protein fibrils extracellular deposit. When there is cardiac involvement, ATTR can cause thickening of the myocardium, impaired diastolic function, and restrictive physiology, which results in heart failure and adverse cardiovascular events. For hereditary ATTR, 2023 ACC expert consensus recommends regular monitoring in asymptomatic gene carriers, starting about 10 years before the proband's onset age followed by testing every 3–5 years unless symptoms appear [44,45,46]. CMR is one of the modalities recommended for the evaluation of suspected cardiac amyloidosis.
CMR is a robust imaging tool to identify the presence, location, and distribution of hypertrophy and detect cardiac amyloid infiltration. A TI-scout (inversion time) determines the optimal nulling time of normal myocardium after gadolinium contrast. Amyloid deposits expand the extracellular space, causing faster gadolinium washout from the blood pool than the myocardium. This reverse nulling pattern has a specificity for cardiac amyloidosis. The pattern of LGE is classically subendocardial or transmural, with very elevated ECV and T1 mapping values providing high sensitivity for amyloid infiltration (Fig. 3). ATTR cardiomyopathy will frequently present with increased LV mass and either low normal or reduced LVEF. The LGE pattern can involve RV and atria. In genetically positive patients, a reduction in circumferential strain can be seen in early amyloid infiltration with high sensitivity.
Fig. 3
Inherited cardiac amyloidosis. Transthyretin amyloid (ATTR) stress cardiac magnetic resonance imaging showed a) moderate left ventricle (LV) concentric hypertrophy (arrow illustrates interventricular septum = 17 mm measured at the short axis (SAX) basal slice at diastole), b) severely reduced LV ejection fraction (LVEF 17%) as illustrated on the long-axis (LAX) four-chamber (4 CH) view at systole; Diffuse late gadolinium enhancement (LGE) in nonischemic pattern as indicated by multiple arrows c) in short axis phase-sensitive inversion recovery (PSIR) LGE image and d) in the four-chamber magnetization-prepared inversion recovery (MagIR), with the latter also showing left atrial LGE. In both views, the nulled blood pool signal can be appreciated as indicated by an asterisk (*); e) abnormal myocardial blood flow (MBF) at rest (0.42 mL/g/min); f) markedly global reduced stress MBF (0.66 mL/g/min) and myocardial perfusion reserve (MPR 1.57). g) Native T1 mapping showed globally increased T1 relaxation time of 1203 ± 5.05 ms (1.5 T) and h) elevated extracellular volume (ECV) fraction (47%)
Elevated ECV and native T1 times are associated with higher amyloid burden, interstitial expansion, and risk of death. In a meta-analysis by Cai et al., each 3% increase in myocardial extracellular volume (ECV) on CMR was associated with a 16% higher mortality risk, and every 60 ms rise in myocardial T1 time corresponds to a 33% increased mortality risk [47].
CMR quantitative perfusion is also abnormal in cardiac amyloidosis. Patients have severely reduced stress MBF and MPR (Fig. 3), correlating with amyloid infiltration, myocardial changes, and disease severity. Therefore, the evaluation of microvascular dysfunction with quantitative CMR perfusion may be valuable for diagnosis, prognosis, and monitoring treatment response [48].
Iron Overload CardiomyopathyIron overload cardiomyopathy can result from primary genetic disorders like hereditary hemochromatosis (four types: HFE, HJV, HAMP, TfR2, and SLC40 A1 mutations), which lead to excessive iron absorption and cardiac accumulation. Secondary causes include autosomal recessive hemoglobinopathies such as thalassemia major (with over 350 mutations on chromosome 11) and sickle cell anemia (which is characterized by a point mutation where valine replaces glutamic acid) as a less common cause. These conditions require repeated blood transfusions, causing iron buildup in the heart. Iron overload induces oxidative stress, mitochondrial dysfunction, and impaired calcium handling, leading to myocardial damage [49].
CMR is the gold standard for detecting myocardial iron accumulation and associated dysfunction. T2* relaxometry measures transverse relaxation time; lower values (< 20 ms) indicate iron overload, with values < 10 ms in severe cases associated with an increased risk of ventricular arrhythmias and heart failure Fig. 4. However, T2* can be affected by motion artifacts and may be less reliable in mild iron overload. T1 mapping offers a more sensitive alternative, measuring longitudinal relaxation time. CMR aids in guiding iron chelation therapy, with T2* values informing treatment response and intensity, while also monitoring disease progression, correlating with LVEF, and detecting early diastolic dysfunction in primary and transfusion-dependent iron overload patients [50, 51, 52].
Fig. 4
Iron overload cardiomyopathy a) Moderately dilated size (end-diastolic volume indexed: 123 ml/m2) and normal wall thickness interventricular septum = 8.5 mm measured at the short axis (SAX) basal slice at diastole) and severely reduced left ventricle systolic function (LVEF: 28%), b) four-chamber (4 Ch) long-axis (LAX) view at systole. There was evidence of mid-wall stripe of late gadolinium enhancement (LGE) in the basal septum and focal epicardial LGE in the basal inferior segment consistent with myocardial fibrosis as illustrated by arrows on the c) short axis and d) four-chamber magnetization-prepared inversion recovery (MgIR) image. e) Native mapping showed globally reduced T1 relaxation time, elevated ECV (42%), and f) reduced T2 relaxation time. g and h) Myocardial T2* map quantification (R2* 94.4 Hz) indicated cardiac iron overload status
Anderson-Fabry CardiomyopathyAnderson-Fabry disease (AFD) is a rare X-linked multisystem genetic disorder with α-galactosidase deficiency leading to abnormal accumulation of glycosphingolipids within lysosomes [53]. Early detection is essential for optimal treatment and to prevent fibrosis in the later stages of the disease. Key CMR features include myocardial hypertrophy, lower T1 mapping values with early glycosphingolipid accumulation, and elevated T2 mapping values with inflammation. CMR detects early changes by measuring an LV global longitudinal and atrial strain before LV hypertrophy or lower T1 values occur. LV hypertrophy and LGE seen on CMR is associated with a higher cardiac event, while their absence suggests a better prognosis [54, 55]. CMR can also be used to monitor treatment response to enzyme replacement therapy. In patients with baseline hypertrophy but minimal LGE, enzyme replacement has been shown to decrease LV wall thickness and mass on CMR. In addition, normalization of T1 and T2 relaxation times correlates with decreasing wall thickness and LV mass. However, enzyme treatment has not been shown to change RV mass and LGE burden [56].
Non-Dilated Left Ventricular Cardiomyopathy (NDLVC)Since 2023, the ESC classified a separate and distinct category of non-dilated left ventricular cardiomyopathy (NDLVC) defined as a cardiomyopathy with non-ischemic LV scarring or fatty replacement without dilation. It may be accompanied by wall motion abnormalities/hypokinesis that is independent of hypertension, valvular disease, or coronary artery disease [17]. It encompasses early-stage DCM, non-dilated hypokinetic forms, and arrhythmogenic LV involvement; however, its prevalence and management are still underexplored. NDLVC shares genetic overlap with DCM and ARVC (Table 1). Mutations can affect severity, progression, and SCD risk. Genetic screening is essential for diagnosis, risk stratification, and family evaluation [17]. CMR is important for diagnosing NDLVC and also for risk stratification, detection of non-ischemic fibrosis, scarring, and adipose replacement. Mid-wall distribution of LGE is a major risk marker and the LGE patterns vary by genetics. T2 mapping identifies edema in inflammatory cases [57]. Eda et al. also found NDLVC in 22% of non-ischemic cardiomyopathy, with these individuals having more AF, less LGE (34% vs 53%), and similar outcomes when compared to DCM [58]. Progression to dilated cardiomyopathy predicted worse prognosis, highlighting the need for serial imaging and risk assessment. A recent retrospective study of 23,472 CMR scans found ring-like LGE in 1.1% of cases. It was a high-risk marker for adverse outcomes and arrhythmias, especially when ≥ 7 segments were involved. This pattern was commonly linked to desmosomal (DSP) and non-desmosomal (FLNC, LMNA) genes with both NDLVC and DCM and less often associated with ARVC, HCM, or other cardiomyopathies [59].
Left Ventricular HypertrabeculationLeft ventricular non-compaction (LVNC), once considered a separate cardiomyopathy, is now recognized as a morphologic trait. Excessive trabeculation can be seen in up to 20% of healthy individuals and in physiologic states such as athletic training or pregnancy [60]. When present with cardiac disease, it often overlaps with other cardiomyopathies, including DCM, NDLVC, ARVC, or other genetic forms (Fig. 5). Over 80 genes have been linked to excessive trabeculation, involving sarcomere structure, mitochondrial function, X-linked disorders, and cardiac development. Hypertrabeculation is also found in 20–30% of patients with Duchenne or Becker muscular dystrophy and is associated with worse outcomes [61]. A non-compacted to compacted (NC/C) myocardial ratio ≥ 2.3 suggests hypertrabeculation is better assessed by CMR and if present the CMR can further, identify underlying phenotypes with specific cardiomyopathy diagnosis. Adverse outcomes are associated with lower LVEF, higher NC/C ratio, enlarged LV volumes, increased mass, fibrosis, and LGE—particularly in genetic variant carriers. CMR-derived atrial strain also helps identify individuals with multiple genetic variants [62].
Fig. 5
Leftventricular hypertrabeculation in dilated cardiomyopathy. a) short axis (SAX) cine image, b) 4 chamber long axis (LAX) cine image at the end of diastole shows dilated left ventricle with highly trabeculated myocardium, which is distributed at the apex and mid anterior and lateral walls (red asterisk). The ratio of non-compaction (red line) and compaction (green line) is > 2.3. c) short axis phase-sensitive inversion recovery (PSIR) late gadolinium enhancement (LGE) image: subendocardial LGE in the mid-lateral wall (yellow arrow)
Peripartum CardiomyopathyPeripartum cardiomyopathy is defined as idiopathic heart failure with a reduced ejection fraction (LVEF < 45%), occurring during the last month of pregnancy or within five months of the postpartum period. Recent studies have shown genetic links in peripartum cardiomyopathy, such as single-nucleotide polymorphisms near the PTHLH gene, along with TTN and BAG3 [
Comments (0)