
Remember me

At the start of this research collaboration, a multidisciplinary team of clinicians, epidemiologists, biostatisticians, data experts, and project managers was formed. The collaborative research team established systematic processes designed to generate meaningful, valid, expedited, and transparent RWE [12, 13], leveraging existing recommendations and collective experience, and developing novel approaches when needed. Our research process consisted of 4 phases: (1) research planning and prioritization, (2) protocol development, (3) protocol implementation, and (4) results dissemination, with operational steps underlying each phase. In Phase 1, we developed a new template to facilitate data source characterization. In Phases 2 and 3, we applied existing study-specific tools (e.g., templates, decision trees, step-by-step processes) to enable decision-grade RWE during study design [14], fit-for-purpose data assessment [15], protocol finalization [14, 16], and protocol implementation [16]. Additionally, through Phase 2, we developed a new study phase diagram to enable communication and transparency of our protocol implementation process.
Phase 1: Research Planning and PrioritizationGenerate Timely EvidenceThe global pandemic necessitated rapid answers to pressing research questions. To allow the research team to implement multiple studies to generate timely evidence, this collaboration used a platform approach to enhance the efficiency, and therefore speed, of analytic implementation. Generally, a platform approach involves use of data with: (a) clear and appropriate data transformations; (b) validated analytic workflows with documentation; (c) reproducible analyses, rather than traditional line-coding; and (d) direct access to underlying assumptions and analytic measures to enable timely evidence generation. Aetion® Substantiate [17] was able to reuse previously built algorithms for defining variables of importance for COVID-related research and the data (where prespecified in protocols) for multiple studies. It also facilitated transparent reporting of all variable algorithms and analytic steps.
Given the rapidly changing clinical care of and epidemiology of COVID-19, the team sought data sources with multiple linked or linkable data types, short lag times (e.g., 1 month lag in time between data capture and analysis ready data,) and frequent refreshes available (e.g., refreshed every 2–4 weeks to add the most recent data), that could be rapidly connected to and analyzed on the analytic platform. Further, the ability to accelerate the contracting process and delivery of the initial data was also considered. While the goal was to provide accurate evidence as quickly as possible, timelines for each study varied considerably due to numerous factors (e.g., complexity, sample size requirements, and number of parallel studies at a given time).
Key Lesson #1: Strategic Data RefreshingFor the data source [20] that was continually refreshed, new data became available generally every two weeks. While data with a short lag time was important for ensuring meaningful evidence—for data explorations, diagnostics, and analyses—refreshing at specific study points (e.g., just before starting the Protocol Implementation) may save effort and time. However, to maintain objectivity, data should only be refreshed prior to study initiation and per protocol.
Systematically Evaluate and Characterize Each Data Source of InterestWhile there were many sources of data being assembled to support COVID-19 research, seven US sources were identified as potential candidates within this project. As a first step, data dictionaries and other written documentation were reviewed (if available). The team then confirmed understanding of essential information such as data provenance and completeness of fields needed for most of the potential research questions (e.g., age, sex) with data providers. For each data source, these elements were compiled into written summaries and organized into a table to effectively communicate key dataset components (e.g., sample size, inclusion criteria, update frequency, type, and level of documentation on source data and transformation) for each data source (Table 2). Given the importance of near real-time data, regular and frequent communication with potential data partners was required to fully understand the composition and nuances of the RWD sources. As an initial step in the process, evaluation of the metadata was needed to assess study feasibility.
Table 2 Standard data characterization templateKey Lesson #2: Establish Structured Data CharacterizationUse of a standard data characterization form – to be completed and regularly updated by data providers – can increase efficiency and speed in this initial phase of work. An example template is shown in Table 2. The intent is to minimize the burden on the data providers prior to contracting, while including enough information to evaluate initial aspects of the data that inform data relevance and reliability [18] and whether the data are appropriate for inclusion in the data feasibility assessment for a specific research question.
Define Initial Research QuestionsPreliminary research questions of interest were identified at the start of the research collaboration through regulatory research priorities, collective idea generation, and review of the relevant literature available. Many of the initial questions focused on characterizing COVID-19 natural history and treatment patterns (Table S1), or on the safety and effectiveness of early treatments (e.g., hydroxychloroquine). For each preliminary research question of interest, specific objectives were defined, and then the research questions were prioritized based on collaborative agreement of current clinical relevance and whether the research question represented a foundational aspect necessary for subsequent assessments (numbers in Table S1 are in order of planned initiation). Research questions were then revised and reprioritized as scientific needs changed over time based on observed treatment patterns (often following emergency use authorization changes) and emerging knowledge from implementation of prior collaboration research questions, other COVID-19 collaborative research efforts such as the COVID-19 Evidence Accelerator [19], and published research.
Key Lesson #3: Prioritize Initial Research QuestionsDeveloping initial research questions allowed prioritization of foundational aspects that were used in subsequent assessments, accelerating implementation. However, flexibility to pivot on downstream research questions was important given learnings from the foundational work and the rapidly evolving health response.
Phase 2: Protocol DevelopmentProtocol development included four operational steps spanning from articulating a specific research question to finalizing the protocol. Protocol development included a step to systematically identify fit-for-purpose data for the intended research question, as well as a step allowing descriptive exploration of the selected data prior to study design and protocol finalization. For full transparency and replicability, all study details, including key study design parameters and operational and implementation details, were recorded using structured templates [14,15,16].
Articulate Research Question(s) and Objective(s)Within the first year of the collaboration, the research team addressed 10 research questions – 9 descriptive and 1 comparative – leveraging RWD to better understand COVID-19, specifically the natural history, potential therapies, and diagnostics (see Table S1) within two US administrative healthcare data sources that comprised claims, hospital chargemaster and electronic health records [20,21,22]. For each study, the research question and objectives were prospectively developed to be sufficiently detailed to ensure alignment among the team and to clearly specify the population and key subgroups of interest, treatment(s), outcome(s), covariates of interest, and timeframe [13, 23, 24].
Key Lesson #4: Prespecify Research Question and ObjectivesArticulating the detailed research question in a structured and comprehensive manner using an existing framework and template [14], facilitated alignment with the research team to ensure laser focus for data source evaluation.
Initiate Real-World Study Design and Identify Fit-For-Purpose DataFor each study, existing frameworks and templates were used to guide design choices, capture the rationale, and identify minimal criteria for a RWD source to meet the needs of the study [12, 14,15,16]. Study design diagrams illustrating key assessment windows (e.g., baseline, exposure, follow-up) were used for all studies to enable study team decision-making and reproducibility. Additionally, for the comparative study [25] the team designed the study as an emulation of a hypothetical pragmatic trial [26, 27] and used Directed Acyclic Graphs [28, 29] to identify potential confounders (care setting, data types, completeness of key data fields, numbers of patients).
Once the minimal data criteria were defined for each study, a data feasibility framework and structured templates were used to guide the data selection process [15]. For data to be fit-for-purpose, data must be both reliable and relevant [30,31,32]. Reliability broadly relates to the accuracy of the data [31, 32], and data relevance pertains to the availability of sufficient patients with key study data elements and representativeness [30, 33]. If a fit-for-purpose data source could not be identified, the research question was revised or not pursued.
Key Lesson #5: Streamline Templates for Study Design and Data Decision RationaleWhile the approach for identifying fit-for-purpose data was considered successful as an adoptable model for future studies, the team felt relevant components of existing reporting templates and required fields [14,15,16] could be merged into one source from the onset as a “master” approach for decision-making. Based in part on this experience, a combined template for study design and data fitness assessment plus specific references to the analytic detail has since been published that may be used to reduce duplication and produce more transparent documentation [34].
Explore Selected Data Source(s)Once the data for a particular research question were accessible (i.e., received, processed, and available on the platform), data were explored to inform protocol decisions on variable definitions and time windows (e.g., the length of the pre-hospitalization baseline period), and identify additional potential threats to validity (e.g., possible misclassification of oxygen support among the group identified as not receiving any supplemental oxygen) and the need for protocol-specified sensitivity analyses to evaluate design decisions and robustness of findings. To maintain objectivity in the design and analysis of each research question, planned data explorations were documented, and any treatment-outcome associations remained blinded per Good Pharmacoepidemiology Practice [35] (e.g., treatments and outcomes were not linked, and treatment-outcome associations were not analyzed or viewed to ensure independence of study design decisions).
The team evaluated and documented the completeness of key variables necessary for defining the minimal criteria such as inclusion/exclusion criteria, subgroups, as well as various algorithms for defining the primary outcome (again, without “unblinding” treatment-outcome associations) to verify data fitness for purpose. General trends of key variables were compared with internal and external benchmarks (as available in existing literature or nationally reported government public health statistics). This proved an important step that highlighted data nuances essential to maximizing validity for each research question/dataset. For example, in the comparative study, this exercise guided the decision to truncate the cohort entry date selection period to end 60 days earlier than the end of the near-real-time data to ensure complete data for reporting the mortality endpoint [25]. The detailed feasibility assessment was refined as data explorations uncovered subtle variations and insights. As during the data feasibility assessment, communication with data partners was required to fully understand the underlying features of the data (e.g., data provenance, data linkage, data transformation), make decisions around adapting variable definitions and the study design, and to ensure transparent acknowledgment of potential limitations.
Once completed, the findings were documented, and all variable definitions or algorithms (how all variables were defined and operationalized in the study), and assessment windows were documented in the structured template [16], as an appendix to the final protocol (see for example, Appendix A in the posted protocol [25]). Key study design decisions derived from the exploratory step were reviewed by all collaborators and incorporated into a final protocol with consensus.
Key Lesson #6: Feasibility Assessment Confirmation Through Data ExplorationData explorations, maintaining blinding, were necessary to yield valid and interpretable study findings. For future studies a newly published process that incorporates diagnostic steps as a standard part of protocol design is available [36].
Develop, Finalize, and Post ProtocolProtocol Development incorporated the key design decisions derived during the exploratory step and concluded with protocol completion. Following published protocol standards and regulatory guidances [35], all study design details were documented in protocols. Protocols included descriptions of the RWD source and fit-for-purpose rationale; study design details, including assessment windows for baseline, exposure, and follow-up (including censoring criteria) [37]; and a statistical analysis plan including primary, secondary, and planned sensitivity analyses. Once protocols were developed for initial research questions, there was an appreciable efficiency gain from using the protocol template and relevant protocol text (e.g., when applying to the same data source, approach and/or study parameters). Visual study design diagrams [37] with assessment windows were also included [25, 38]. For the comparative study, graphical depictions of potential confounders and potential threats to internal validity (e.g., misclassification, incomplete data, and residual confounding) were proactively identified, and the protocol specified how these limitations would be addressed in the study design and analysis.
Protocols specified phased implementation with (1) built-in diagnostic tests and objective criteria to be met before conducting the analyses (e.g., specific requirements for baseline covariate balance), (2) analytic contingencies tied to each diagnostic test, and (3) required team consensus to move to each subsequent implementation phase [25]. Similar to the data exploration step, these protocolized diagnostics and criteria were carefully specified to avoid unblinding any treatment-outcome association to maintain objectivity (i.e., to ensure that implementation decisions were unbiased). We developed a protocol template aligned with published standards and guidance, enabling efficiency and consistency in our documentation. However, there is now a protocol template designed to harmonize protocol development best practices and guidance, and incorporate templated study analytic details [16, 37, 39] that were not available for consideration in this study.
Existing guidelines unanimously agree on the importance of posting final protocols for non-interventional (observational) studies. Posting final protocols for such observational studies on a publicly available website (such as ClinicalTrials.gov or ENCePP.eu) prior to study implementation provides transparency. Public posting is one way that might provide external confidence in the prespecified design and allows evaluation of whether the implementation was influenced by the findings [32], integral for studies intended to support causal inferences [40]. Therefore, the final approved protocol for the comparative study was publicly posted on ClinicalTrials.gov [25]. All studies were also covered under an IRB exemption determination from New England IRB.
Key Lesson #7: Protocol DesignIncorporation of phased study conduct into the protocol design helped us remain blinded to treatment-outcome associations (to maintain objectivity) during the design and baseline diagnostic phases of the comparative study. However, there are three interrelated aspects that could be incorporated into future studies, particularly when clinical care for the disease or condition of interest is rapidly evolving.
Treatment Patterns: Treatment pattern findings from the exploration phase did not signify the potential for post-baseline treatment changes, and therefore adjustment for potential informative censoring was not included in the comparative protocol. However, treatment patterns drastically changed over the period from Protocol Development to Protocol Implementation. These changes, combined with our strict adherence to the protocolized phased approach (which was specified to ensure objectivity), created challenges in the comparative study. Because evaluation of post-baseline data until the inferential phase was not allowed, the substantial treatment crossover that occurred just after the study index date was not known and it was not feasible to amend the protocol prior to the inferential phase. Thus, inverse probability of censoring weighting was used to evaluate the impact of this crossover on the hazard ratios post hoc [41]. The need to reanalyze changing treatment patterns prior to beginning the inferential phase was a lesson learned through this research.
Analytic Contingencies: As noted above, due to the drastic treatment changes, some design and analysis decisions made based on initial data explorations were not fully aligned to the actual study data. To address this in future work, we would add objective diagnostic criteria for post-baseline treatment changes to identify potential informative shifts in the treatment paradigm prior to the inferential phase. Analytic contingencies would be prespecified that correspond to any suboptimal diagnostic criteria triggering adjustment for informative censoring, if warranted. Although it is difficult to anticipate every possible change in patient care or the research environment, it is crucial we build in analytic contingencies to address major changes in clinical care (such as potential channeling towards or away from study treatments over time) that would otherwise impact target trial emulation [42, 43]. Prespecified contingency plans may also allow revision of the research question if the study is likely to have reduced interpretability or relevance due to changes in standards of care.
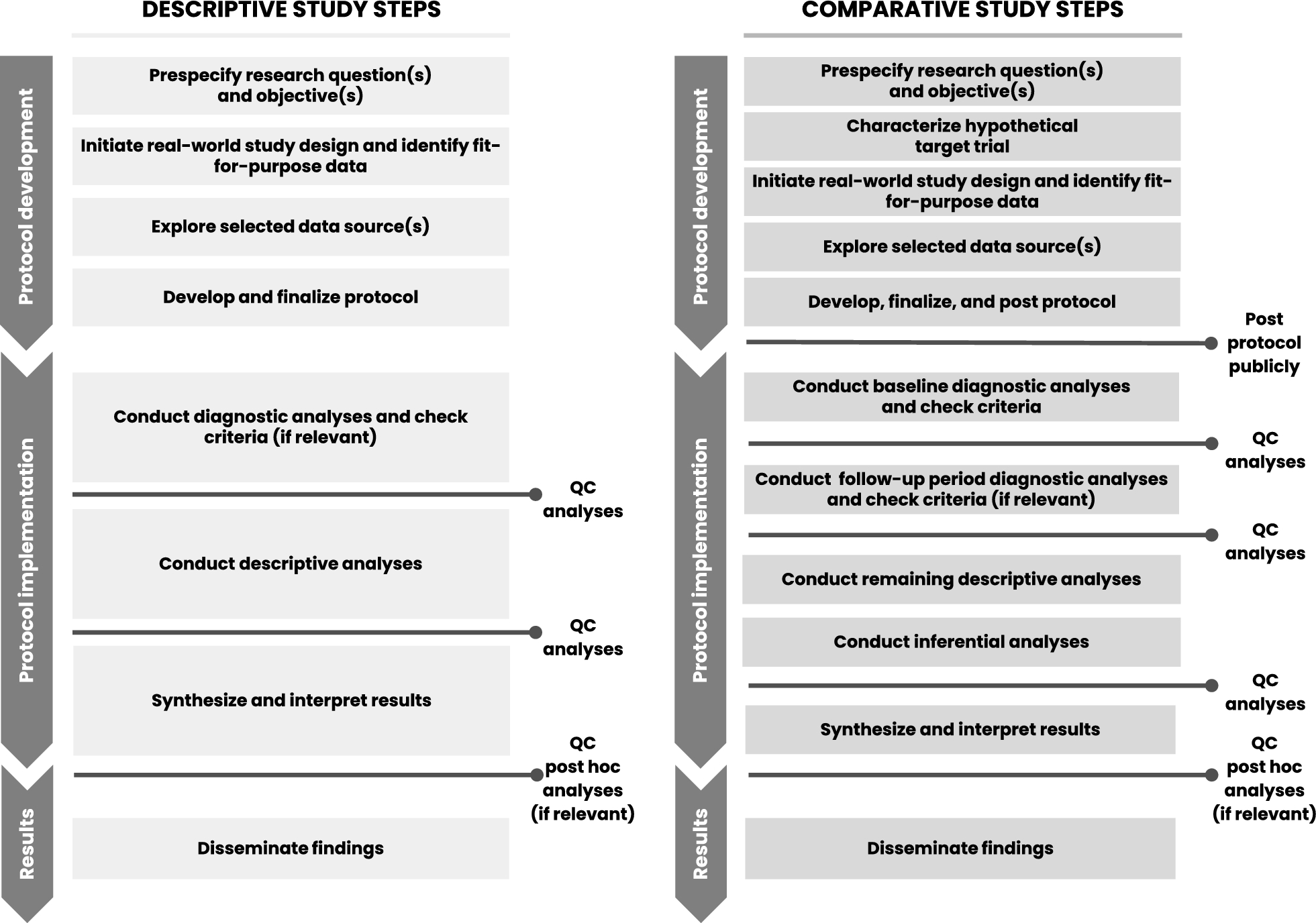
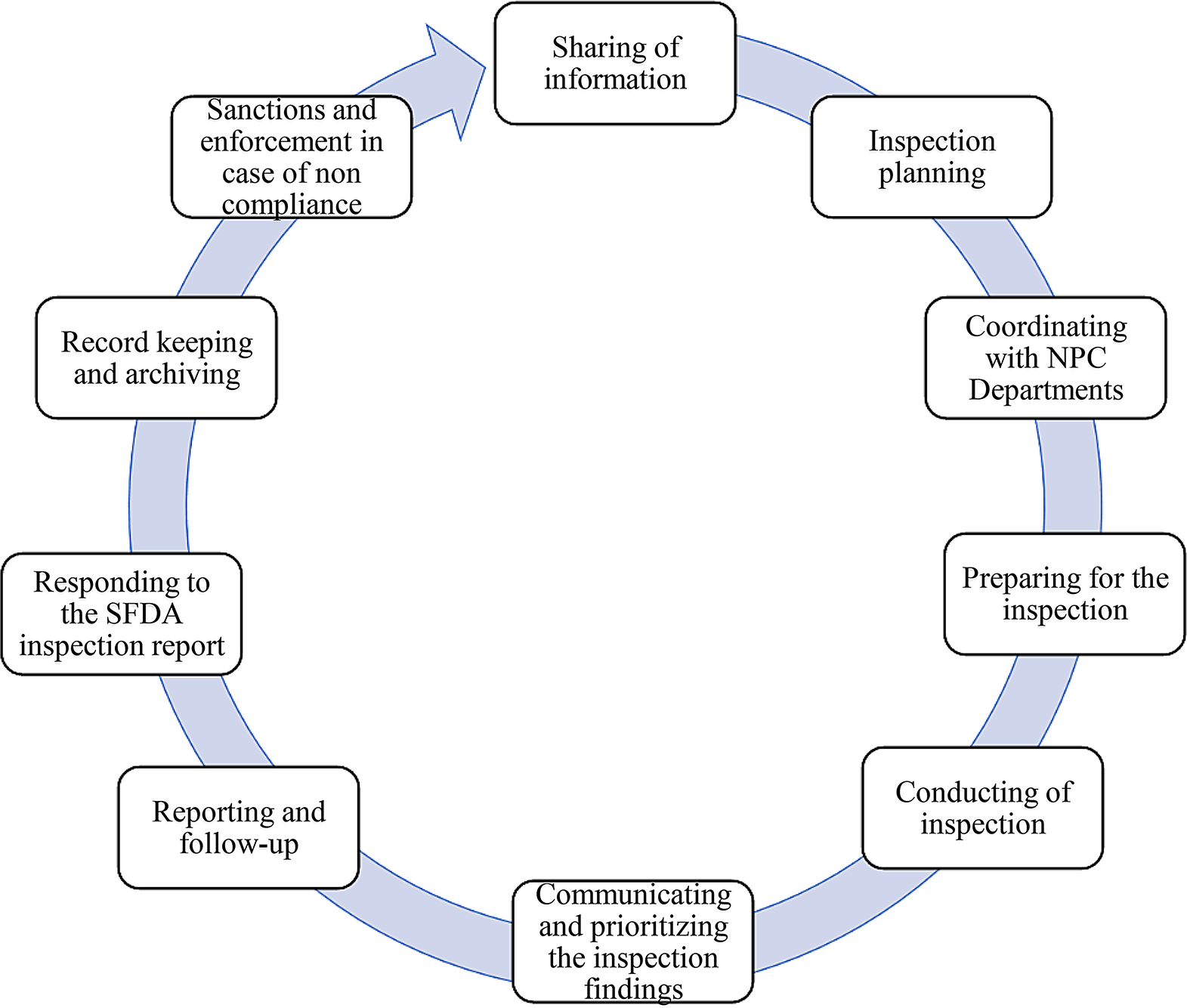
Diagnostic Criteria: To evaluate and analyze informative treatment changes in prespecified analyses, objective diagnostic criteria are needed for post-baseline treatment changes. Future comparative study protocols may consider a second phase diagnostic step that could include evaluation of post-baseline treatment patterns, crossover, and reasons for censoring. Figure 1 provides a proposed phased study process that incorporates the two diagnostic phases for comparative studies—one for baseline data diagnostics and a second for follow-up period diagnostics. However, the analysis of post-baseline data requires careful consideration to ensure outcome blinding of treatment-specific endpoints to support decision making. Thus, this second diagnostic phase would be implemented only after the first (baseline) diagnostic phase is complete to ensure implementation objectivity. Like the first phase diagnostics, the protocol should specify objective criteria to be met before initiating inferential analyses and/or should link diagnostics findings to specific contingent analyses. Having this second diagnostic phase would also allow the research team to consider amending the protocol prior to inferential implementation, if warranted by an unanticipated change that would otherwise reduce the interpretability of the study findings. In fact, these learnings were applied and further developed in a methodologically focused evaluative study [44, 45].
Figure 1
Proposed process for descriptive and comparative effectiveness studies with designated checkpoints required to proceed.
Phase 3: Protocol ImplementationConduct Diagnostic Analyses and Check CriteriaFor all research questions, we implemented protocols as planned, starting with the prespecified diagnostic step. If study diagnostic criteria were not met initially, planned contingencies and iterations were implemented until all criteria could be satisfied. This included waiting for data refreshes if a sample size criterion was not met or consolidating or removing parameters of the propensity score model to satisfy the positivity assumption. Study diagnostic findings were reviewed by all key collaboration members, and findings and decisions were recorded [16].
Key Lesson #8: Prespecified Analysis ImplementationEvaluating diagnostic findings and recording decisions further enabled transparency and efficient presentation and publication, and minimized the number of post hoc analyses conducted.
Conduct Descriptive Analyses (and Inferential, if Relevant)After study diagnostic criteria were reviewed with consensus from the team, any remaining descriptive analyses were implemented, followed by inferential analyses. Implementation findings and decisions, along with the rationale, were documented [16].
Synthesize and Interpret ResultsOnce all planned protocol analyses were completed and quality control processes were applied, data were synthesized and reviewed by all collaboration members. Post hoc analyses were identified by consensus and documented in the structured template [16]. Post hoc analytic plans were then created and implemented. Of note, these analyses may not be appropriate for decision making and may have limited interpretability.
Key Lesson #9: Post Hoc Analysis ImplementationConsidering post hoc analyses only after all prespecified analyses were complete and reviewed allowed focused identification of key additional analyses to further aid in the interpretation of primary prespecified results. Documenting the objective and potential interpretation of post hoc analyses prior to implementation provided clear rationale to justify additional analyses and how they diverged from the prespecified analyses, and further enabled team alignment on these additional analytic steps.
Phase 4: Results DisseminationDisseminate FindingsFor all treatment-specific research questions, final results were shared publicly through peer-reviewed conference abstracts and presentations (n = 7), in FDA Science Forums (n = 3), and as manuscripts (n = 5) (Table S1). Presentations and publications focused on prespecified analyses, and clearly delineated all post hoc findings when presented. The dissemination of these efforts supported understanding of treatment-related COVID-19 research, methodological approaches to ongoing regulatory science research efforts across centers to answer public health questions, as well as informing the broader clinical and scientific community.
Key Lesson #10: Peer-review PublicationWhile results were shared as quickly as was feasible and conference submissions were an efficient way to disseminate results relatively rapidly, manuscript submissions to journals presented challenges, as clinical and epidemiologic journals were inundated with COVID-19 submissions after the first year of the pandemic. Use of “brief report” style papers rather than full length manuscripts covering all primary and secondary analyses may have resulted in more timely manuscript peer review and publication. Additionally, exploring systems to better accommodate the need for peer review or provide a mechanism to incentivize such review to meet necessary public health challenges is worth evaluation.
Comments (0)