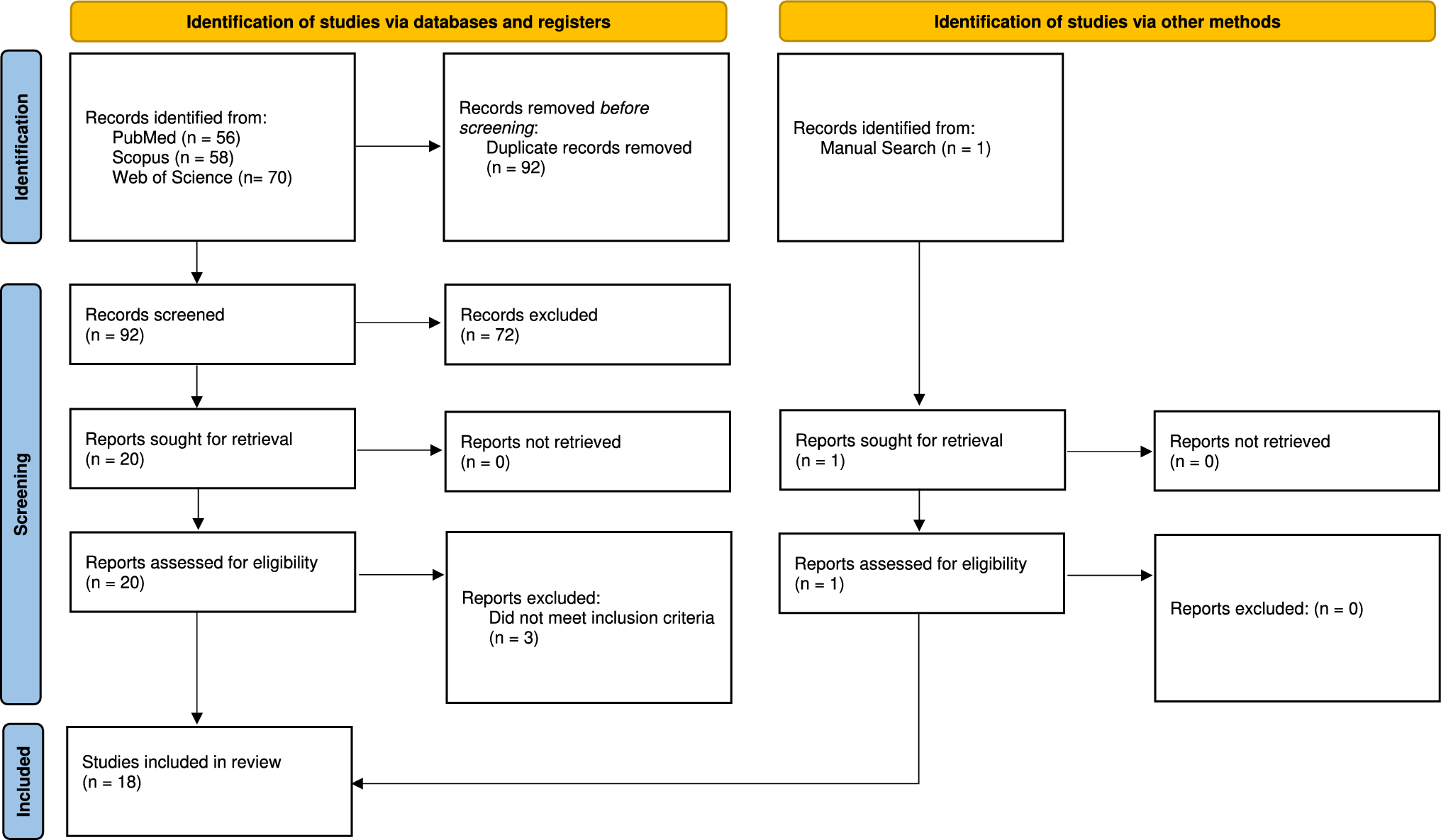
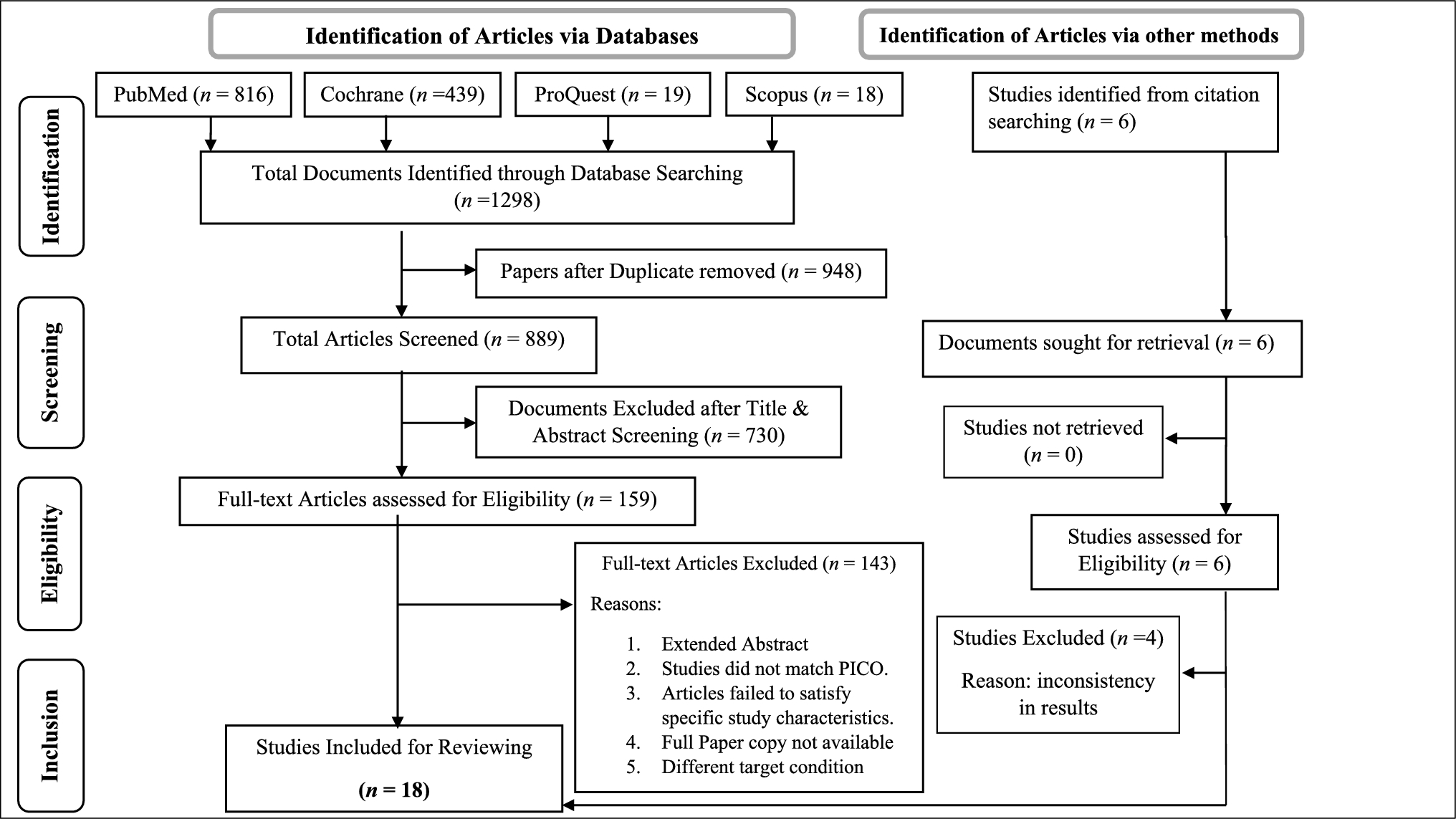
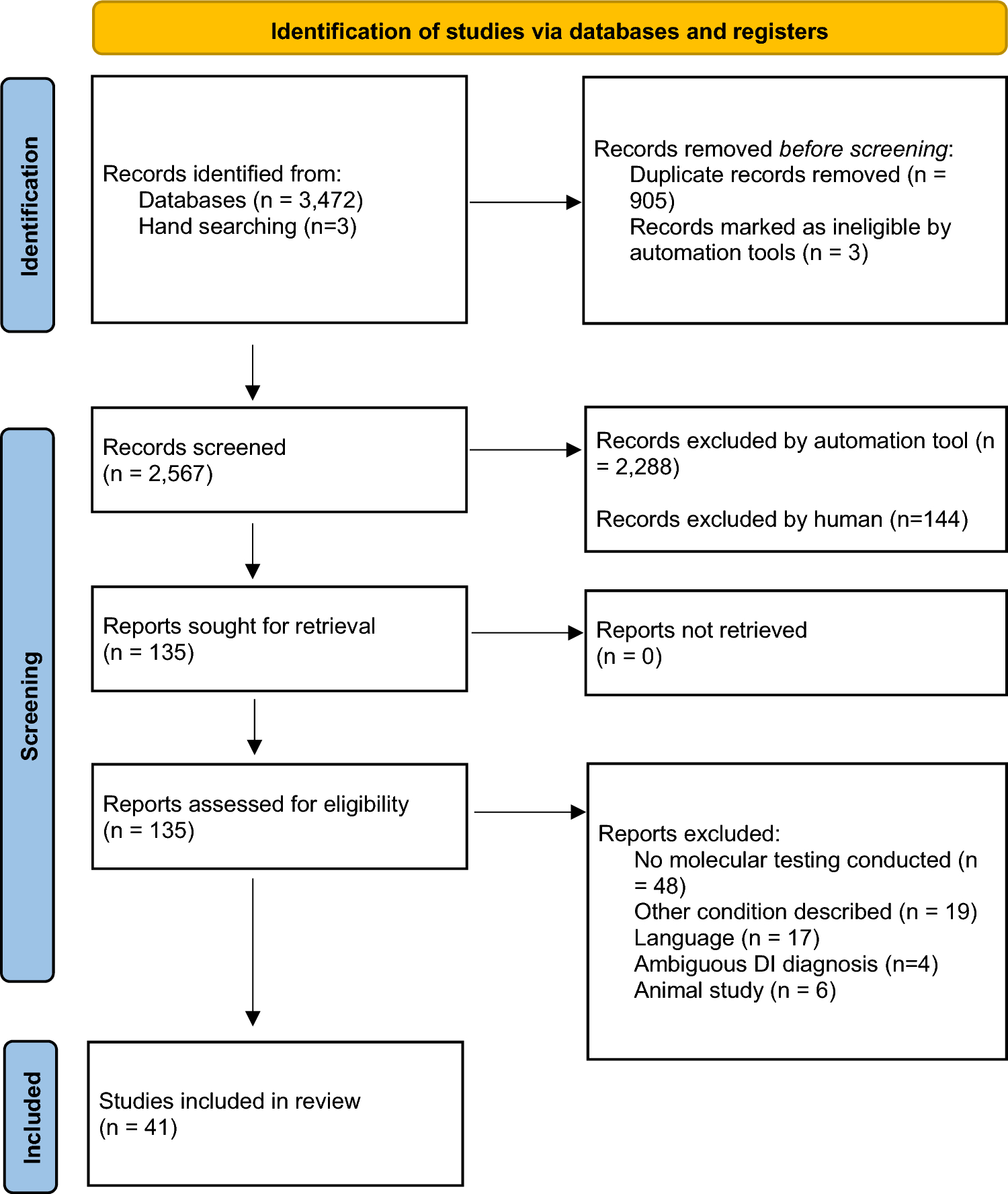
The objective of this study was to consolidate the existing molecular data related to DI as reported in the literature. Within the scope of this research, we identified and reviewed a total of 322 documented cases of individuals with DI.
Mutations within the DSPP gene have a profound impact on the development of both enamel and dentine, as DSPP is expressed by odontoblasts and temporarily in pre-ameloblasts (Bègue-Kirn et al. 1998). The DSPP gene is composed of five exons and four introns and encodes a precursor protein that undergoes cleavage, resulting in the production of two mature proteins: DSP and DPP. Exons 1–4 are responsible for encoding DSP, while exon 5 encodes both the carboxy terminus of DSP and DPP. DSP and DPP play vital roles as non-collagenous components within dentine and are crucial for the mineralization process. Research involving mutant mice has demonstrated that DSP primarily regulates the initiation of dentine mineralization, whereas DPP is associated with the maturation phase of dentine mineralization (Suzuki et al. 2009).
Mutations occurring outside the DSPP gene have been documented in a limited number of cases involving non-syndromic DI, specifically in COL1A1 and COL1A2. These genes are responsible for encoding type I collagen, the primary structural component of pre-dentine, comprising approximately 90% of its composition. Beyond its pivotal role in dentine, type I collagen is a fundamental component in various tissues, including bone, tendons, and skin. Consequently, mutations in COL1A1 and COL1A2 can result in dentine anomalies seen in syndromes like OI. However, recent research findings have identified mutations in the COL1A1 and COL1A2 genes in patients with non-syndromic DI, devoid of any OI-related characteristics. These discoveries indicate that these genes may serve as notable mutational hotspots associated with non-syndromic DI.
In Table 1, the mutations span different exons and introns, leading to a range of protein alterations and disease manifestations. For instance, mutations in Exon 2 include c.16 T > G (p.Y6D) and c.44C > T (p.A15V), with reported cases numbering 7 and 8, respectively, and are associated with developmental disorders (DD-II) and developmental impairments (DI-II). Notably, the c.50C > T mutation (p.P17L) is prevalent with 18 cases and consistently linked to DI-II, as highlighted in several studies including those by Li et al. (2012) and Simmer et al. (2022). Intron 2 mutations, such as c.52-3C > G and c.52-25del23bp, also demonstrate significant variability in phenotypic outcomes. The c.52-2A > G mutation, causing p.V18_Q45del, affects two individuals with DI-III and DI-II, reflecting a diverse range of developmental impairments. Mutations in Exon 3, such as c.133C > T (p.Q45X) and c.53 T > A (p.V18D), are also well-documented with 18 affected individuals respectively and are associated with developmental impairments of both DI-II and DI-III types. Exon 5 mutations, including c.1918_1921delTCAG and c.2525delG, predominantly result in developmental disorders (DD-II and DI-II), with multiple studies documenting these variants in various populations. The data reveal that Exon 2 mutations, primarily missense types, affect individuals across American, Asian, and European populations, with a notable prevalence in Asia. Intron 2 mutations, predominantly splice-site changes, are most common in European populations, with significant representation in Asia as well. Exon 3 mutations, including both missense and nonsense types, are predominantly found in Asian individuals. Intron 3 mutations, all splice-site variants, are heavily concentrated in Asia. Exon 5 mutations, including frameshift and insertion/deletion types, affect a broad range of populations, indicating widespread genetic impact across continents. A single case of a c.G1463C mutation in Exon 22, resulting in a p.G488A protein change, has been reported. This missense mutation was observed in an Asian individual and is associated with a developmental impairment phenotype classified as DI-I. The findings are detailed in the study by Zeng et al. (Zeng et al. 2021) The data reveal several mutations found in Asian populations with specific regional distributions. The c.1009G > A mutation in Exon 19 (p.Gly337Ser), linked to developmental impairment (DI), was observed in 2 cases in Thailand (Udomchaiprasertkul et al. 2020). The c.1171G > A mutation in Exon 21 (p.Gly391Ser), associated with developmental impairment type I (DI-I), was reported in 9 individuals from Thailand and South Korea (Kantaputra et al. 2018; Lee et al. 2021). The c.3233G > A mutation in Exon 48 (p.Gly1078Asp) was noted in 3 cases from South Korea though its specific phenotype is not categorized (Lee et al. 2021). These findings provide a clearer understanding of the mutation distributions and their phenotypic associations in different Asian regions.
Mutations in genes other than DSPP, COL1A1 and COL1A2 may cause non-syndromic DI. However, such genetic mutations are yet to be uncovered. In these cases, WES is an ideal genetic testing approach to identify causative mutations in other genes. The relationship between specific genetic mutations and their phenotypic manifestations in DI is complex and crucial for both clinical management and research. The DSPP gene, which encodes the DSPP, plays a central role in dentine formation. Mutations in DSPP lead to various forms of DI, each with distinct clinical and histological features. For instance, in cases of Exon 2, mutation predominantly associated with DI-II, impacted the protein structure, and disrupted dentine matrix formation, leading to softer and more brittle dentine. Clinically, patients with these mutations may experience severe tooth discoloration and fragility, necessitating proactive dental care and intervention. Similarly, Exon 5 mutation leads to frameshift changes, often resulting in severe DI forms. These mutations are linked to significant structural abnormalities in dentine, which are observed as irregular dentinal tubules and compromised mineralization. Clinicians should anticipate these severe manifestations and prepare for more intensive dental management strategies. The identification of non-DSPP mutations in COL1A1 and COL1A2 suggests a broader genetic basis for DI beyond DSPP. For example, mutations in COL1A1 and COL1A2 can result in dentine anomalies similar to those seen in OI. This finding underscores the importance of considering a broader genetic screening panel when diagnosing DI, particularly in patients with atypical presentations. Non-DSPP DI, namely COL1A1 and COL1A2 mutations, can exhibit a broader range of phenotypic presentations, sometimes overlapping with OI symptoms, but without the full clinical spectrum of OI. Histopathological differences, such as variations in pulp chamber morphology and the extent of pulp obliteration, further distinguish non-DSPP cases from DSPP-related DI.
Microhardness test results indicated that the dentine hardness of DI teeth was lower than that of healthy teeth. This decrease in hardness can be attributed to DSPP mutations, which result in the formation of dentine with structural and hardness defects. Scanning electron microscopy results revealed teeth with reduced and irregular tubules. Energy-dispersive X-ray spectroscopy analyses (Du et al. 2021, 2023 and Zeng et al. 2021) showed changes in phosphorus, magnesium, sodium and calcium compared to healthy controls, confirming impaired mineralization of DI dentine. The abnormalities in dentinal structure and elemental content often lead to premature tooth fractures and periapical infections. Abnormal pulp morphology can further complicate dental treatment. Therefore, comprehensive, long-term dental management is of paramount importance for individuals with DI.
The higher portion of Asians could be due to both geographic and sampling tendencies. Geographically, certain DI mutations may be more prevalent in Asian populations, aligning with regional genetic variability. Additionally, enhanced diagnostic capabilities and awareness in Asian countries might lead to more frequent detection and reporting of DI cases. Sampling tendencies could also play a role as majority of the studies included in the review are from Asian regions or if there's a publication bias favoring studies from these areas. To fully understand this discrepancy, it's essential to examine both regional prevalence data and potential biases in study selection and reporting. The histological differences according to genotype in DI highlight how specific genetic mutations impact the dentine structure and formation. DSPP-related DI typically shows more consistent and severe histological abnormalities, while non-DSPP DI (especially with COL1A1 and COL1A2 mutations) may present with a broader range of histological variations and additional features related to systemic conditions like OI.
The critical missing element in the current studies is the detailed connection between genotype and phenotype, specifically how mutations in different exons of the DSPP gene manifest both clinically and histologically. While the studies provide valuable information on the types of mutations present in various exons, they often fall short of correlating these genetic variations with specific clinical features and histopathological findings. For instance, a mutation in Exon 2 impacts the dentine’s structure such as tubule morphology or mineral density. However, the translation of these findings to clinical symptoms, such as tooth discoloration or sensitivity, remains unexplained. Without this direct linkage, clinicians lack crucial insights into how these mutations affect disease progression and presentation, impeding the development of targeted diagnostic and therapeutic approaches. Addressing this gap requires more integrated studies that correlate genetic data with detailed clinical and histological outcomes.
Shields’ classification remains a staple in clinical practice due to its established framework, ease of use, and direct applicability. While de La Dure-Molla’s classification provides valuable insights into the genetic aspects of DI, Shields' system offers a more practical approach for everyday clinical decision-making (de La Dure-Molla et. al. 2015, Shields et al. 1973). Among the 41 studies included, 37 followed Shield’s classification, 3 were ambiguous, and 1 utilized the Modified Shield’s classification.
The current study has its limitations. The varying criteria classifying DI across included studies, can impact the comparability of results and the overall synthesis of data. The reliance on studies published in English and the exclusion of experimental studies might limit the diversity of data, potentially skewing results. Furthermore, the inclusion of only well-documented cases with clear DI classification could result in the omission of relevant studies with less definitive diagnoses. The data predominantly focus on studies from specific regions, which may not fully represent global genetic diversity. The concentration of data from Asian populations, for example, might introduce regional biases in understanding mutation prevalence and phenotypic expressions. While the chosen methodology provides a comprehensive overview of DI-related genetic variants, the inherent limitations of classification variability, study biases, and regional focus must be considered. Future research should aim to standardize classification criteria, include diverse populations, and leverage advanced genetic sequencing techniques to enhance the understanding of DI and improve patient management strategies.
Additionally, understanding the genotype–phenotype interactions in DI is critical for advancing clinical practice and improving patient care. By integrating comprehensive genetic testing into clinical practice, utilizing advanced sequencing technologies, and addressing the limitations of current studies, clinicians can enhance diagnosis, prognosis, and management of DI. The accumulated variant data analysis in this systematic review provides a further basis for increasing our comprehension to better predict the occurrence of DI and appropriate patient management. Future research should focus on elucidating the molecular mechanisms of DI, expanding genetic screening, and standardizing diagnostic criteria to improve patient outcomes and guide personalized treatment approaches particularly those related to the expression of DSPP, COL1A1 and COL1A2.

Comments (0)