
Remember me



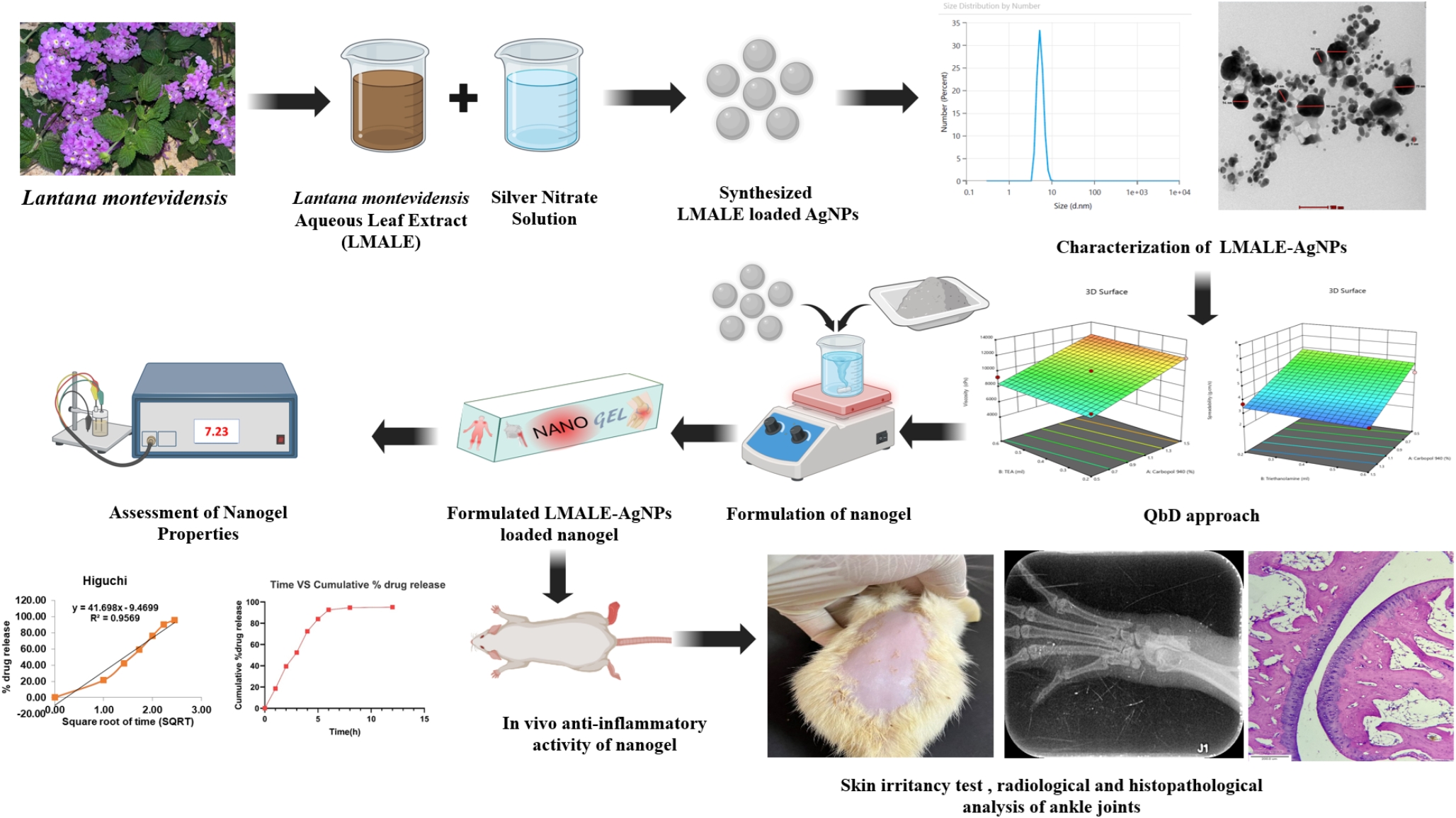
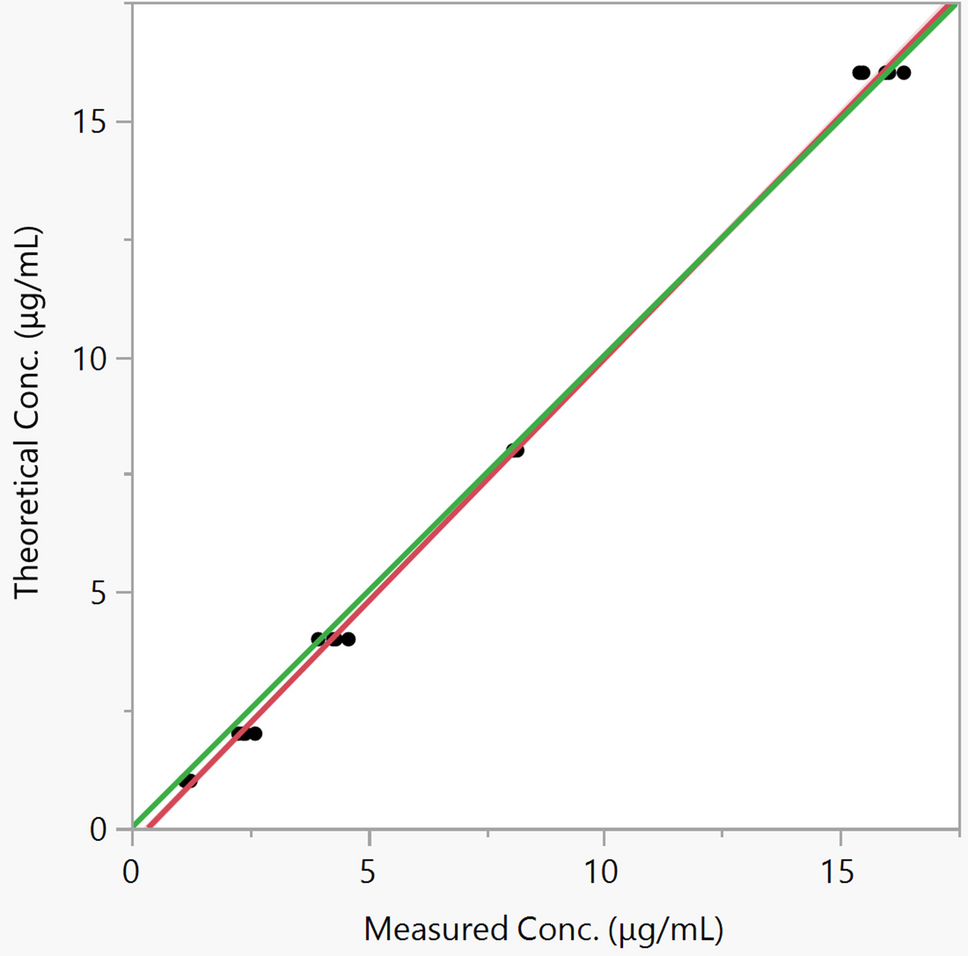
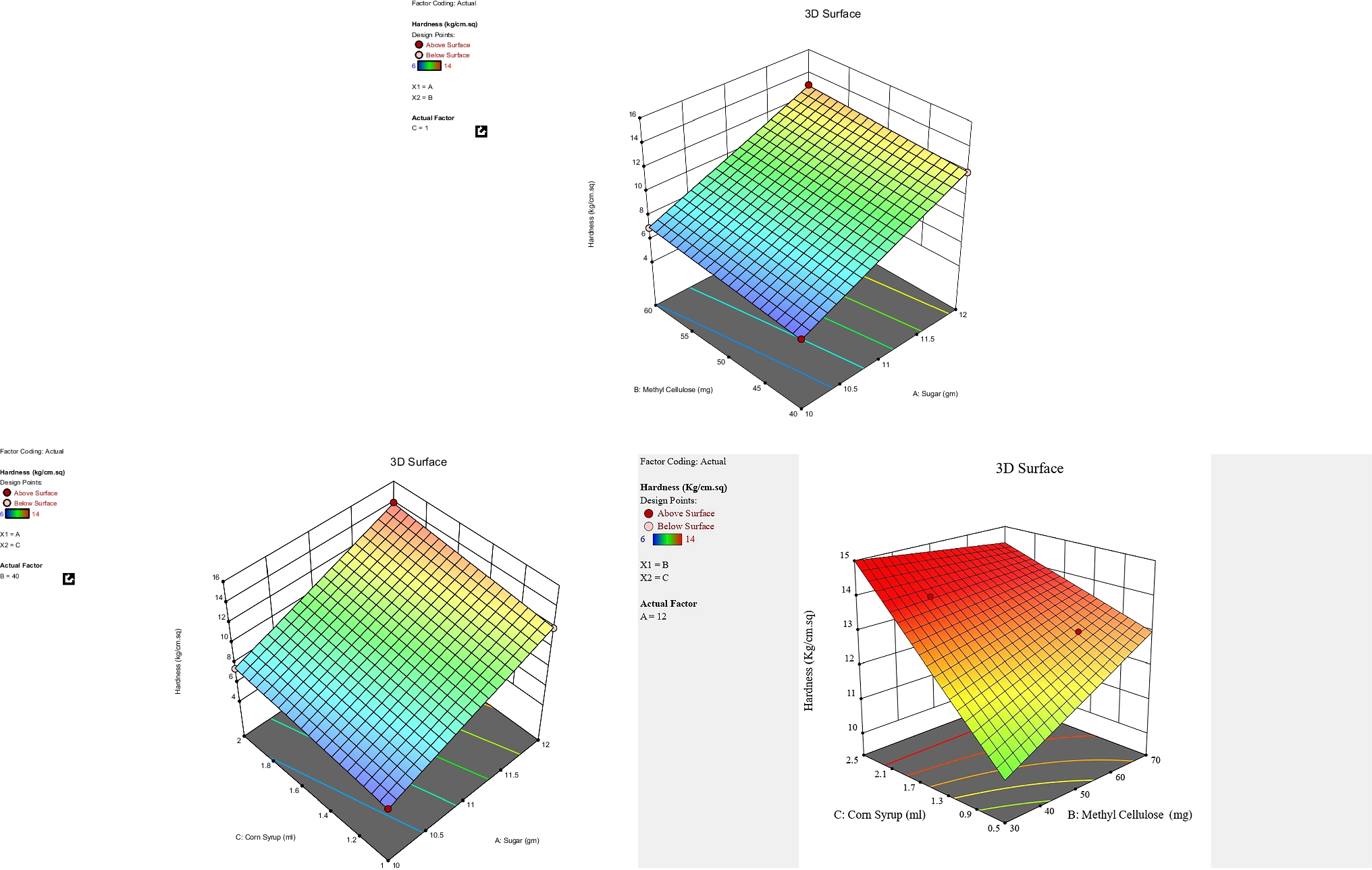
A total of 269 accelerated approvals were granted from December 11, 1992, to June 30, 2021 [3], and 175 (65%) were in oncology (Fig. 2). The 175 accelerated approvals included 18 with variations in dosing and administration (pembrolizumab and larotrectinib). After excluding these 18 accelerated approvals, 157 accelerated approvals were analyzed. The indications for these accelerated approvals were distributed across 39 cancer types. Of these, non-small-cell lung cancer (18/157, 11%), breast cancer (13/157, 8.3%), and chronic myeloid leukemia (11/157, 7.0%) were the three major indications for which the accelerated approval pathway could be leveraged for expedited market access. In addition, four accelerated approvals with specific gene profiles were indicated for cancer-agnostic solid tumors: pembrolizumab (tumor mutational burden-high; microsatellite instability-high or mismatch repair deficient), entrectinib (neurotrophic tyrosine receptor kinase gene fusion), and larotrectinib (neurotrophic tyrosine receptor kinase gene fusion).
Fig. 2
Flow for sample selection
Characteristics of Clinical Evidence Submitted for Accelerated ApprovalThe clinical evidence submitted for accelerated approval is summarized in Table 1. Hundred twenty accelerated approvals (76%) were based upon one trial. When focusing on the features of clinical trials leading to accelerated approval, 124 accelerated approvals (79%) were based on non-comparative single- or multi-arm trial(s) (Table 2). In addition, 141 accelerated approvals (90%) were based on the response rate results. Hundred forty-seven accelerated approvals (94%) reported response rate, either as a primary or secondary endpoint, with response rates ranging 8–80% (median, 42.9%; not presented in tables). The median duration of response was reported for 119 accelerated approvals. For 88 products that reached the median, with the median duration of response ranging 3.8–21.7 months (median 9.6, months).
Table 1 Number of pre- and post-accelerated approval clinical trials for efficacy for oncologyTable 2 Primary endpoint of clinical trial leading to accelerated approval and the most robust primary endpoint for post-accelerated approval clinical trials for oncologyFDA Rationale for Granting Accelerated ApprovalThe FDA rationale for granting accelerated approval is summarized in Supplement 2. Sixty accelerated approvals (38%) were based on “favorable benefit-risk profiles” without referring to other existing products, and 26 accelerated approvals (17%) were because the FDA concluded “superior efficacy/safety over existing therapy” was shown. Of these 26 accelerated approvals, only 6 presented the results of a comparative trial for accelerated approval, and the remainder referred to historical evidence. Notably, for 13 accelerated approvals (8%) the FDA determined that the efficacy and safety requirements for regular approval were not met and accelerated approval was granted instead. The rationale was not identified in 39 accelerated approvals (25%) because neither FDA review reports nor alternative publications were available.
Association Between the Accelerated Approval Rationale and Prior Regulatory DesignationProducts granted accelerated approval were often given prior regulatory designation(s), such as orphan drug (105/157, 67%), fast track (40/157, 25%), breakthrough therapy (56/157, 36%), and priority review (87/157, 55%) (Supplement 2). When looking at accelerated approvals with these “favorable” and “acceptable” benefit-risk profiles, accelerated approvals because of “acceptable benefit-risk profiles” accompanied with prior fast track, breakthrough therapy, or priority review designation more often (17/19, 89%) than accelerated approvals with “favorable benefit-risk profiles” (44/60, 73%).
Post-Accelerated Approval Requirements/Commitments for Regular ApprovalUpon granting the accelerated approval, 232 post-accelerated approval requirements were issued by the FDA for 157 accelerated approvals, accompanied by 189 post-marketing requirements and 284 post-marketing commitments, 162 of which required reporting (Supplement 3). Generally, the post-accelerated approval requirements stipulated the execution of supplemental trials and follow-up of pre-accelerated approval trials (218/232, 94%), whereas the post-marketing requirements often advised investigating efficacy and safety in special populations, such as pediatrics and patients with hepatic or renal impairment (54/189, 29%) and drug-drug interactions (41/189, 22%). Post-marketing commitments with reporting requirements often described supplemental clinical evidence (58/162, 36%), drug-drug interactions (25/162, 15%), and device development (25/162, 15%). Other post-marketing commitments were mainly CMC tests (107/122, 88%; Supplement 3).
In almost all accelerated approvals (155/157, 99%), one or more supplemental clinical trials or a follow-up of the clinical trial submitted for accelerated approval were required or committed to be conducted in accordance with either post-accelerated approval requirement, post-marketing requirement, or post-marketing commitment where the clinical trial with the most robust study design was stipulated in the post-accelerated approval requirement (Note; OS-RCT was deemed more robust than TTE-RCT), although in two cases (i.e., dabrafenib and trametinib) an OS-RCT was conducted spontaneously in accordance with the post-marketing commitment and another supplemental TTE-RCT was required as the post-accelerated approval requirement.
The Number of Pre- and Post-Accelerated Approval TrialsMore than half of the oncology accelerated approvals (84/157, 54%) were granted based on one trial, and an additional trial was required for conversion to regular approval (Table 1). For 21 accelerated approvals (13%), one trial accounted for the approval and additional 2 trials were either required or committed for conversion.
Poisson regression analysis showed that the ratio of supplemental trials to the total number of trials expected for regular approval was smaller when more pre-accelerated approval trials were presented (coefficient = -1.625, p < 0.001), and when the effect size over standard of care was larger (-0.776, p < 0.001) (Table 3, Model A). A coefficient of -1.625 indicated that an increase in the number of pre-accelerated approval trials in one trial resulted in a decrease in the number of supplemental trials required by the FDA by approximately 20% (i.e., exp (-1.625)). In another model that considered only explanatory variables with p values of < 0.1, prior breakthrough therapy designation was associated with more supplemental trials (0.346, p = 0.039; Model B). Regarding indications, fewer supplemental trials were required when products were indicated for respiratory (-0.465, p = 0.035) and female cancers (-0.532, p = 0.016) than for hematologic cancers.
Table 3 Factors that may have characterized the number of post-accelerated approval clinical trials for oncologyPrimary Endpoint of Pre- and Post-Accelerated Approval TrialsWith respect to the primary endpoint of the pre- and post-accelerated approval trials, 57 of 157 accelerated approvals (36%) were based on single- or multiple-arm non-comparative trials (s) with the primary endpoint of response rate, followed by additional TTE-RCT(s) for conversion to regular approval (Table 2). Another 33 accelerated approvals (21%) were based on single- or multiple-arm non-comparative trials (s) with the primary endpoint of response rate, followed by an additional OS-RCT.
The results of the ordered logistic regression analysis are presented in Table 4. The primary endpoint for post-accelerated approval trials tended to be more robust when accelerated approval was granted for more common cancers (coefficient = 0.336, p = 0.020), but less robust when more subjects were treated with the products in the pre-accelerated approval trial(s) (-0.596, p = 0.002), when accelerated approval was granted for respiratory cancers (-1.531, p = 0.069), skin cancers (-2.437, p = 0.007) or “other” types of cancers (-2.001, p = 0.029), and when cancers with higher 5-year survival rate were targeted (-2.855, p = 0.007; Model C). In another model that considered only the explanatory variables with p values of < 0.1, favorable benefit-risk profiles may have allowed a less robust confirmatory trial in the context of the primary endpoint (-0.641, p = 0.088; Model D). In addition, the findings were similar to those of the previous model.
Table 4 Factors that may have characterized the most robust primary endpoint of post-accelerated approval clinical trials for oncology
Comments (0)