
Remember me


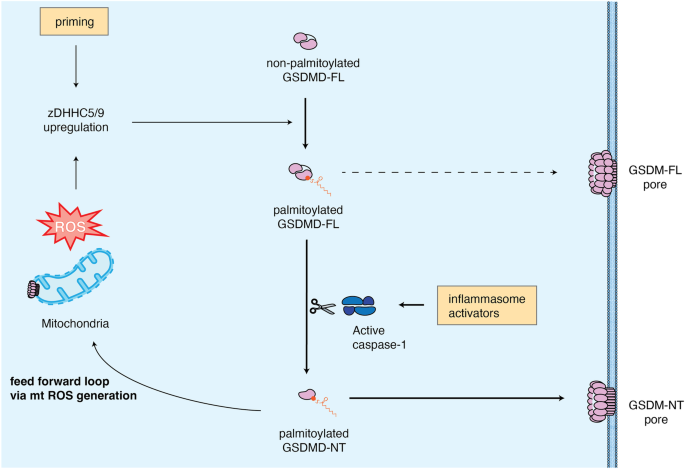

Gasdermin D (GSDMD) is the pore-forming executor of pyroptosis and cytokine secretion during inflammasome activation. In a recent study in Nature, Du et al. report that S-palmitoylation of human GSDMD at Cys191 (mouse Cys192) is a key regulatory step that controls both its activation and interaction with membranes.
Palmitoylation is a reversible post-translational modification (PTM) of proteins involving the addition of a 16-carbon fatty acid, palmitic acid, by cellular palmitoyltransferases (PATs). Palmitoylation commonly occurs on cysteine residues (S-palmitoylation), and less frequently on serine and threonine residues (O-palmitoylation). Palmitoylation most commonly affects membrane localization of proteins, although it can also affect protein stability.
Gasdermins (GSDMs) are a family of pore-forming proteins that control the induction of lytic cell death known as pyroptosis.1 While initially identified in mammals, recent studies found that GSDMs also control death in bacteria and fungi.2 Mammalian GSDMs share a domain architecture consisting of a C-terminal (CT) domain that binds and autoinhibits a cytotoxic N-terminal (NT) domain.1 Cleavage of the interdomain linker by proteases relieves autoinhibition, allowing the GSDM-NT to target cellular membranes, where it forms transmembrane pores (Fig. 1). The best-studied family member is GSDMD, which executes pyroptosis downstream of inflammasome complexes, multi-protein signaling hubs that are assembled during the innate immune response to pathogens and sterile insults. In humans, inflammasomes control the activation of caspase-1 and caspase-4, which process GSDMD at Asp275. Following its cleavage, GSDMD-NT exposes basic amino acids that allow binding of acidic phospholipids such as phosphoinositide phosphates, phosphatidylserine, phosphatidic acid and cardiolipin, and lead to association with target membranes. GSDMD-NT preferentially targets mitochondria and the plasma membrane, where it forms β-barrel pores having 31–34-fold symmetry and an internal diameter of 21.5 nm.3 GSDM pores promote the release of proinflammatory cytokines like IL-1β and induce cell lysis by activating ninjurin-1, which perforates the plasma membrane.
Fig. 1: Regulation of GSDMD activity and pore formation by palmitoylation.
At steady state, palmitoylation of GSDMD enhances its association with membranes and GSDMD pore formation upon inflammasome-driven caspase-1 activation. A feed-forward loop via the formation of GSDMD pores in mitochondria induces ROS, which promotes further GSDMD palmitoylation. Since palmitoylation weakens autoinhibition of GSDMD, it can — under conditions of strong ROS production — also result in the formation of pores composed of GSDMD-FL.
Previous studies found that GSDMD oligomerization and pore formation is enhanced by elevated levels of cellular reactive oxygen species (ROS) and that oxidation of GSDMD Cys191 (Cys192 in mouse) plays a major role.4 Interestingly, Cys191 was already well-known to play a critical role in GSDMD pore formation as its mutation reduced GSDMD oligomerization and cell death. Furthermore, the same residue was found to be the target of several GSDMD inhibitors (dimethyl fumarate, necrosulfonamide and disulfiram) that either bind or modify Cys191.2 This led to the hypothesis that this cysteine might stabilize GSDMD pores by forming disulfide bonds. However, since the recent cryo-EM structure of GSDMD pores showed that this was sterically impossible,3 Du et al.5 hypothesized that S-palmitoylation could explain the importance of GSDMD Cys191.
In their study, the authors used alkyne-azide click chemistry and acyl-biotin exchange (ABE) to show that both full-length (FL) GSDMD and GSDMD-NT expressed in HEK293T cells were S-palmitoylated, which could be reduced by 2-bromopalmitate (2-BP), a nonspecific palmitoylation inhibitor. To identify the palmitoylated residue, the authors mutated all cysteine residues in the protein individually and found that only the C191A mutation abrogated palmitoylation. To test the functional relevance of C191 palmitoylation, the authors removed palmitate using ABE and found that it reduced GSDMD oligomerization. Liposome permeabilization assays further demonstrated that GSDMD purified from mammalian cells induced faster pore formation than Escherichia coli-expressed GSDMD (bacteria lack PATs and therefore largely lack palmitoylation) and that de-palmitoylation by hydroxylamine treatment markedly reduced liposome leakage.
Next, the authors investigated whether GSDMD is palmitoylated upon nigericin-induced activation of the NLRP3 inflammasome in human THP-1 macrophages or mouse immortalized bone marrow-derived macrophages. Interestingly, they found that while priming of cells with LPS upregulated GSDMD palmitoylation, LPS and nigericin were necessary to induce full palmitoylation of GSDMD, which depended on C191/C192. Since ROS was shown to enhance GSDMD palmitoylation, the study also tested the impact of mitochondrial complex I inhibitor rotenone (ROT) and complex III inhibitor antimycin A (AMA) that elevate ROS production. Both ROT and AMA increase palmitoylation of GSDMD and cell death in HEK293T cells or THP-1 macrophages, which could be reduced by ROS scavengers or 2-BP. Intriguingly, GSDMD deficiency reduced ROS production upon inflammasome activation, indicating that ROS production requires GSDMD pore formation. Since reconstitution with C191A GSDMD or the reduction of mitochondrial cardiolipin exposure by knockout of either cardiolipin synthase 1 or phospholipid scramblase 3, reduced ROS production, the data support a model in which palmitoylated GSDMD drives mitochondrial ROS production and thus further palmitoylation in a feed-forward loop (Fig. 1).
While testing the impact of C191 on GSDMD-induced cell death and IL-1β, the authors noticed that GSDMD-D275A, the cleavage-deficient form used as a negative control, exhibited low-level cytotoxic activity and also caused ROS production. Moreover, incubation of liposomes with palmitoylated wild-type or D275A GSDMD still caused leakage, albeit much reduced relative to palmitoylated GSDMD-NT. Negative stain microscopy of these liposomes revealed 30-nm-diameter pore-like structures, similar to GSDMD-NT pores, indicating that GSDMD-FL can form pores when palmitoylated. A cryo-EM structure at 5.5 Å resolution showed that palmitoylated GSDMD (D275A)-FL pores had a 33-subunit stoichiometry similar to the GSDM-NT pores, but featured additional density, probably representing the GSDMD-CT, on the same plane as the transmembrane region. It is conceivable that upon association with a membrane, the GSDMD-CT assumes a different conformation thereby allowing insertion of the GSDMD-NT into the membrane. Interestingly, the cryo-EM structure of palmitoylated monomeric GSDMD showed that palmitoylation weakened the interaction between GSDMD-NT and -CT, further supporting that GSDMD-FL could be activated independently of cleavage. Such a mechanism could be relevant for the autism-associated V41A mutant of GSDMD that weakens autoinhibition. Expression of GSDMD V41A was found to enhance ROS production and GSDMD palmitoylation, suggesting that spontaneous pyroptosis caused by GSDMD V41A is cleavage-independent and driven by GSDMD-FL pores.
Finally, the authors tested which of the 23 human zinc-finger DHHC (zDHHC) motif-containing PATs was responsible for GSDMD palmitoylation and found that zDHHC5 and zDHHC9 interacted with GSDMD and that their absence decreased GSDMD palmitoylation and inflammasome-induced cell death. Of note, a second study published concomitantly6 also found that zDHHC5 and zDHHC9 drive GSDMD palmitoylation at Cys191/192. Interestingly, the authors of this study found that priming was the main driver of GSDMD palmitoylation, and that palmitoylation directed the membrane translocation of GSDMD-NT but not of GSDMD-FL. Thus, while both studies agree on the importance of S-palmitoylation for GSDMD-NT pore formation, additional work is needed to fully characterize whether pore formation by GSDMD-FL occurs under all conditions.
The discovery that ROS-dependent S-palmitoylation acts as a key regulatory step for GSDMD activation has an important impact on our understanding of GSDM regulation and activation. The finding explains not only the role of Cys191/192 in pore formation, but also the mechanism by which dimethyl fumarate, necrosulfonamide and disulfiram inhibit GSDMD. Moreover, the fact that a PTM can weaken autoinhibition and thus allow pore formation by GSDMD-FL has important implications, as it demonstrates that cleavage might not always be necessary to trigger GSDM activation. Indeed, a recent study showed that certain fungal GSDMs do not need to be cleaved to be activated.7 For example, Trichoplax adhaerens features a GSDM that is kept autoinhibited as a disulfide-bonded dimer and activated to form pores after reduction of disulfides. Another example is the regulator of cell death-1 (Rcd-1) protein in the filamentous fungus Neurospora crassa, a GSDM homolog consisting only of the pore-forming domain, which forms heteromeric pores responsible for allorecognition in Neurospora.
Given this new insight into GSDM biology, it will be important to investigate whether other mammalian GSDMs can be activated by PTMs or other cleavage-independent mechanisms, under which physiological settings this occurs, and how this is linked to metabolism and the redox-state of host cells.
Comments (0)