
Remember me
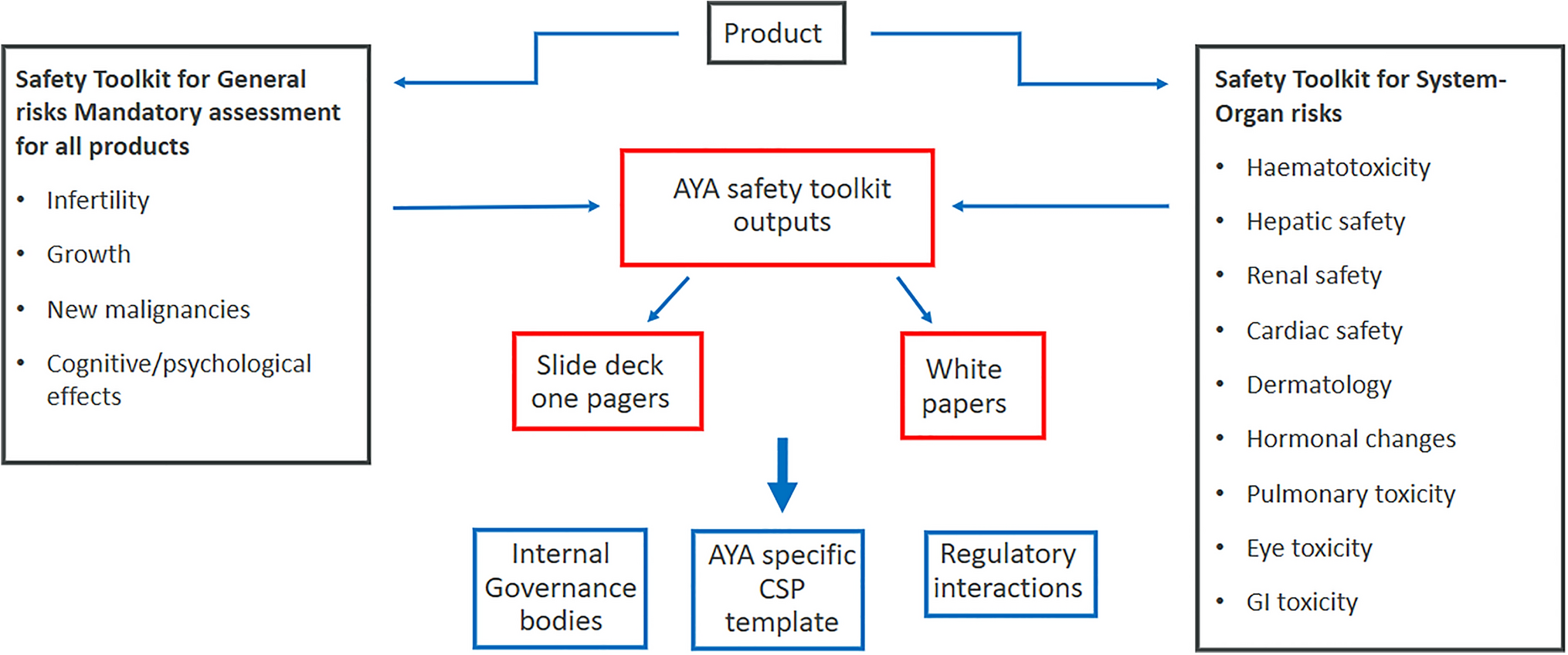

Over the years, the FDA has recognized the need to incorporate clinically meaningful data into prescribing information for already-approved drugs. The FDA’s emphasis on safety and optimization is highlighted in the dose optimization cases of cabazitaxel and ceritinib (see Supplementary Information). Cabazitaxel was first approved on June 17, 2010. The FDA issued 2 PMRs for additional data that compared the approved dose with a lower dose to reduce hematological toxic effects and infections. On September 14, 2017, the FDA issued a supplemental approval based on the fulfillment of the PMRs, which lowered the recommended dose of cabazitaxel from 25 mg/m2 to 20 mg/m2. Similarly, for ceritinib, a KI approved on April 29, 2014, the FDA required a study comparing different doses in fasted versus fed states. On December 21, 2017, study results resulted in a label change from 750 mg with no food within 2 h and ceritinib discontinuation for patients unable to tolerate 300 mg with food, to a new label with a reduced dose of 450 mg with food and ceritinib discontinuation for patients unable to tolerate 150 mg with food.
With 16%, or 1 in 6, of NMEs approved from 2016 to 2022 having a dose-related PMC/PMR, the FDA has balanced the need to expeditiously review and approve novel therapies for patients with cancer with the uncertainty around optimal dose at the time of approval. There tends to be greater scrutiny in the postmarketing setting for novel therapies as they generally have less certainty around their safety profile and less long-term follow-up.
It is not surprising that a greater proportion (15 of 21; 71.4%) of dosing-related PMCs/PMRs from 2016 to 2022 were issued to KIs and ADCs/PDCs (Fig. 1, Table 2). KIs are well known for having off-target effects, particularly non-specific KIs that inhibit multiple kinase pathways simultaneously [10, 11]. These off-target effects may not only cause acute toxicities but also chronic AEs that may adversely affect the patient’s quality of life and result in the patient’s discontinuation of the therapy over time [10, 11]. Many ADCs/PDCs also have tolerability issues due to their narrow therapeutic index. These toxic effects often lead to skipped doses, dose reductions, or discontinuations due to the limited number of dosing cycles patients can tolerate [12, 13]. The FDA has made it clear that it is no longer to only assess DLTs when considering safety concerns, but sponsors must also assess low-grade, chronic toxic effects that may decrease patient adherence or drug tolerability [1, 2, 4, 5, 7].
There has been a generally increasing frequency of PMCs/PMRs related to dose optimization over the years (Fig. 2, Table 4), from 5.3% in 2019 to 10% in 2022 (Table 2). Between 2016 and 2019, 3 out of the 8 (37.5%) dose-related PMCs/PMRs that have been fulfilled with relevant, supplemental published labels have resulted in labeling changes to their USPI. The noticeable increase of approvals with PMCs/PMRs in 2021 and 2022 may be attributed to KIs and ADCs, as 50% of KIs and 40% of ADCs from 2016 to 2022 received a dose-related PMC/PMR in 2021 and 2022 alone (Fig. 1). Moreover, Project Optimus was introduced in 2021, suggesting a stronger actionable commitment toward dosing optimization in the premarketing setting by the FDA, which may have also contributed to the recent increase in PMCs/PMRs.
Fig. 2
Label changes associated with dose-optimized PMCs/PMRs for oncology therapies from 2010 to 2022. Relevant label changes include changes to dose strength, frequency, modifications, safety, and/or efficacy. Not applicable includes no label found around the PMC/PMR fulfillment date, product withdrawal, or pending study completion
Table 4 Outcomes of PMCs/PMRs for Oncology Therapy Indications from 2010 to 2022In January 2023, the FDA posted a draft dose optimization guidance that emphasized the need to collect the appropriate data regarding the dose–exposure response and other pertinent pharmacokinetic/pharmacodynamic (PK/PD) information to comprehensively assess the benefits versus risks for efficacy and safety/tolerability when developing new oncology drug and biological products [7]. Since novel oncologic therapies are changing the treatment landscape, the FDA expects sponsors to reassess the traditional approach toward dose-finding studies. Alternatives include more comprehensive dose-finding studies through multiple cohorts or expansions, using simulations and models, and a deeper understanding of the patient’s PK/PD to mitigate the risk of unnecessary toxic effects over multiple dosing cycles [2, 5, 7, 14].
One of the primary concerns from outside stakeholders with the FDA’s dose optimization guidance from Project Optimus is that enrolling additional patients to multiple dose levels without first demonstrating the therapy’s clinical activity will subject too many patients to ineffective agents while exposing them to unnecessary toxicities [15]. In addition, industry stakeholders have submitted comments to the FDA’s guidance highlighting the potential risk of exposing patients with life-threatening diseases to a subtherapeutic dosage, indicating a need for flexibility when recommending randomized studies of multiple dosages [15]. This debate indicates that there are some challenges ahead regarding managing speed, uncertainty, and patient risk–benefit profile to appropriately optimize a drug’s dose. Nonetheless, it is the new guidance around Project Optimus that will lead to more rigor and potentially longer timelines and larger dose-finding programs in the premarketing setting.
There were some limitations to this study. We could not confirm some of the PMC/PMR fulfillment dates due to potentially incomplete publicly sourced data, and therefore, the results might not comprehensively represent the landscape of dose-related PMCs/PMRs over the years. In addition, the exclusion of PMCs in special populations (e.g., by ethnicity, hepatic/renal insufficiency, or drug–drug interaction) might underrepresent the number of dose-related PMCs/PMRs for a particular drug. Because these populations may require modified doses or may be uniquely susceptible to dose adjustments, their inclusion in this analysis may artificially inflate the dose-related labeling changes associated with the primary indication population.
Overall, the effectiveness and flexibility of the FDA’s proposed dose selection strategies remain to be seen. FDA’s recommended dose optimization strategies have shown the potential to further evolve the drug development paradigm with the risk of extended premarketing drug development timelines, especially for therapies with newer mechanisms of action. Future studies will need to assess whether the increased timelines and rigor in the premarketing setting will reduce the number of postmarketing dose-related studies leading to relevant labeling changes.
Comments (0)