Tissue harvest and cell isolation
HFP tissue was obtained from 4 osteoarthritic patients undergoing total joint replacement surgery and 1 patient undergoing knee arthroscopy after obtaining informed consent under approval by the ethics committee from Karl Landsteiner Private University (EK 1020/2020). Cell isolation was performed as previously described [20, 25]. Briefly, HFP tissue was minced into 5 mm pieces with a scalpel and digested with 9000 units of collagenase I (Sigma-Aldrich, #C0130) per 10 g of HFP in 15 ml DMEM (GIBCO, #21969-035) for 2 h at 37 °C in a 50 ml tube on a roller. Afterwards, the suspension was filtered through a cell strainer (Corning, #431,750), cells were washed once in PBS and resuspended in growth medium containing DMEM, high glucose, GlutaMAX™ and pyruvate (GIBCO, #31,966,047), 2% Penicillin/Streptomycin (Sigma-Aldrich, #P4333), 1% Amphotericin B (Sigma-Aldrich, #A2942), 10% heat inactivated fetal calf serum (GIBCO, #11,550,356 ), 1% nonessential amino acids (GIBCO, #11,140,050) and 1 ng/ml bFGF (Sigma-Aldrich, #F0291). Cells were seeded at a density of 10,000 cells/cm2 in T175 flasks for expansion culture at 37 °C/5% CO2. Human articular cartilage was obtained from one male patient undergoing joint replacement surgery at the Universitätsklinikum Krems. Informed consent was obtained from the patient. Chondrocytes were isolated and cultured as previously described (De Luna-Preitschopf et al., 0.2017).
Preparation of blood products CPRP and hypACT
Whole blood was collected in-house from 5 to 7 donors chosen randomly from a pool of volunteers after an informed consent was signed. Blood collection was approved by the local Ethics Committee of the Danube University Krems (ESC1020/2020). Inclusion criteria for blood donation were age between 25 and 45 as well as being healthy on the day of blood donation according to an evaluation form asking for conditions such as pregnancy, underweight or diabetes as exclusion criteria. Citrate anticoagulated PRP (CPRP) was generated via collecting whole blood into citrate-coated vacutainer tubes (VACUETTE 9NC trisodium citrate 3.2%, Greiner BioOne, #455,322) and further processed as previously described [20]. hypACT was prepared via reconstitution of freeze-dried hypACT serum powder (kindly provided by Orthosera GmbH) with 2 ml sterile double distilled water.
Metabolic activity via XTT assay
To determine metabolic activity after cell isolation, 1000 cells per well were seeded in 96 well plates in triplicates, with separate wells for each time point up to 14 days. To monitor metabolic activity in the VWBR, an aliquot of 250 µl was drawn from the cell suspension and 20 µl suspension was added to 80 µl growth medium per well in 96-well plates in triplicates. After adding 50 µl XTT reagent prepared according to the manufacturer’s recommendations (Roche, #11,465,015,001) per well and incubation for 4 h at 37 °C / 5% CO2, absorbance was measured at 492 nm with 690 nm correction on a Synergy 2 plate reader. A blank was prepared for each measurement by adding XTT reagent to medium only.
Differentiation of isolated MSCs
Tri-lineage differentiation was performed for 5 donors. Cells were cultured in growth medium or differentiation media as described below supplemented with either 10% CPRP, 10% hypACT or 10% FCS as control. As CPRP required addition of 2 U/ml heparin (Gilvasan Pharma GmbH, #3,909,969) into the culture medium, all conditions were performed in presence of heparin.
Adipogenic differentiation
The adipogenic differentiation potential was determined via Oil red O staining as previously described [20]. In brief, HFP-MSCs were seeded into 6 well plates at a density of 2*105 cells and were cultured in normal growth medium until 100% confluency for 3–5 days, before differentiation was started. Differentiation medium was prepared from serum free growth medium containing StemXVivo® Adipogenic Supplement (Bio-Techne Ltd., Abingdon, United Kingdom). It was further supplemented with either 10% CPRP, 10% hypACT or 10% FCS as control and was changed every 3–4 days. After 21 days, cells were rinsed with PBS, fixed in 10% formalin for 30 min and stained with Oil Red O (Sigma, #01391) according to the manufacturer’s recommendations. Fixed cells were washed with 2 ml distilled water, before 1 ml 60% isopropanol was added for 2 min. Afterwards, 1 ml Oil Red O working reagent (3 parts Oil Red O stock solution, 2 parts distilled water) was added for 5 min. Then, excess dye was gently washed away with distilled water until the water stayed clear. Stained cells were visualised using a phase contrast microscope. Additionally, incorporated dye was extracted via 300 µl 100% isopropanol, centrifuged at 14,000 x g for 5 min and absorbance was measured for 80 µl supernatant in triplicates at 490 nm on a Synergy 2 plate reader.
Chondrogenic differentiation
The chondrogenic differentiation potential was determined via chondropellet formation as previously described [7]. Briefly, 2.5*10^5 cells were mixed with chondrogenic medium composed of DMEM high glucose (GIBCO, #10,596,010), 1% insulin-transferrin-selenium (ITS) liquid media supplement (Sigma-Aldrich, #I3146), 100 nM dexamethasone (Sigma-Aldrich, #D4902), 50 µg/ml ascorbic acid (Sigma.Aldrich, #A4403), 1% non-essential amino acids (GIBCO, #11,140,050), 5 ng/ml TGFβ-3 (PeproTech, #AF-100-36E), and 4% methylcellulose (Sigma-Aldrich, #M7027) and were centrifuged at 4164 x g for 10 min. Pellets were cultured for 21 days at 37 °C/5% CO2 in 15 ml tubes with loose cap and media were changed twice a week. Then, pellets were recovered from the culture medium and excess liquid was removed. Pellets were put on base molds (Fisher Scientific, #22-363-554) prepared with a layer of frozen tissue matrix (Tissue-Tek® O.C.T.™, Sakura Finetek, #4583) and put at -80 °C for at least 24 h. Cryosectioning was performed on a cryostat device (Cryostar NX70, Thermo Fisher Scientific) and 6 μm thick slices were placed on adhesive glass slides (Thermo Scientific™ SuperFrost Plus™, Thermo Fisher Scientific, #10,149,870). Slides were dried and fixed in cold acetone (-20 °C) for 10 min. Histologic staining was performed with Alcian Blue 8GX (Sigma-Aldrich, #A3157) prepared from 1 g Alcian Blue 8GX, 97 ml distilled water and 3 ml 96% acetic acid (Sigma-Aldrich, #A9967) for 30 min. Sections were washed in running tap water for 1 min, dehydrated through 95% ethanol and 2 changes of absolute ethanol for 3 min each. Slides were mounted with xylene before adding a cover glass. Sulfated glycosaminoglycans appeared blue and were visualised in a light microscope to determine the diameter of the chondropellets.
Osteogenic differentiation
The osteogenic differentiation potential was determined via Alizarin S staining as previously described [25]. In brief, HFP-MSCs were seeded into 6 well plates at a density of 2*10^5 cells and were cultured in normal growth medium until 100% confluency for 3–5 days, before differentiation was started. Differentiation medium (serum free growth medium supplemented with 100 nM dexamethasone (Sigma-Aldrich, #D4902), 50 µg/ml ascorbic acid (Sigma-Aldrich, #A4403) and 10mM β-glycerol phosphate (Sigma-Aldrich, #G9422)) supplemented with either 10% CPRP, 10% hypACT or 10% FCS as control were changed twice a week. After 21 days, cells were rinsed with PBS, fixed in 10% formalin for 30 min and stained with aqueous 2% Alizarin Red S solution (Sigma-Aldrich, # A5533), pH 4.3, via covering the cells with 1 ml dye solution for 30 min at room temperature. Afterwards, cells were rinsed with distilled water 3 times and mineralized areas appearing red were visualized via light microscopy. Then, stained cells were incubated with 500 µl 10% acetic acid for 30 min, collected with a cell scraper, vortexed and heated at 85 °C for 10 min, cooled down, and centrifuged at 20,000×g for 15 min. The supernatant was neutralized with 500 µl 10% ammonium hydroxide and absorbance was measured for 100 µl in triplicates at 405 nm with a Synergy 2 plate reader.
Flow cytometry
HFP-MSCs adhering to cell culture dish 48 h post isolation or after EV collection 48 h after switching the cultures to serum free medium were used for flow cytometry employing the StemFlow kit (BD, #562,245). 105 cells were stained with 4 µl positive or negative antibody cocktail and 1 µl compensation control antibodies were used. The positive markers (CD90, CD105, CD73 and CD44) and negative markers (CD34, CD11b, CD19, CD45, HLA-DR) were involved in the analysis. Data acquisition and analysis was performed with a CytoFlex device and FlowJo software vX.0.7, respectively.
EV production
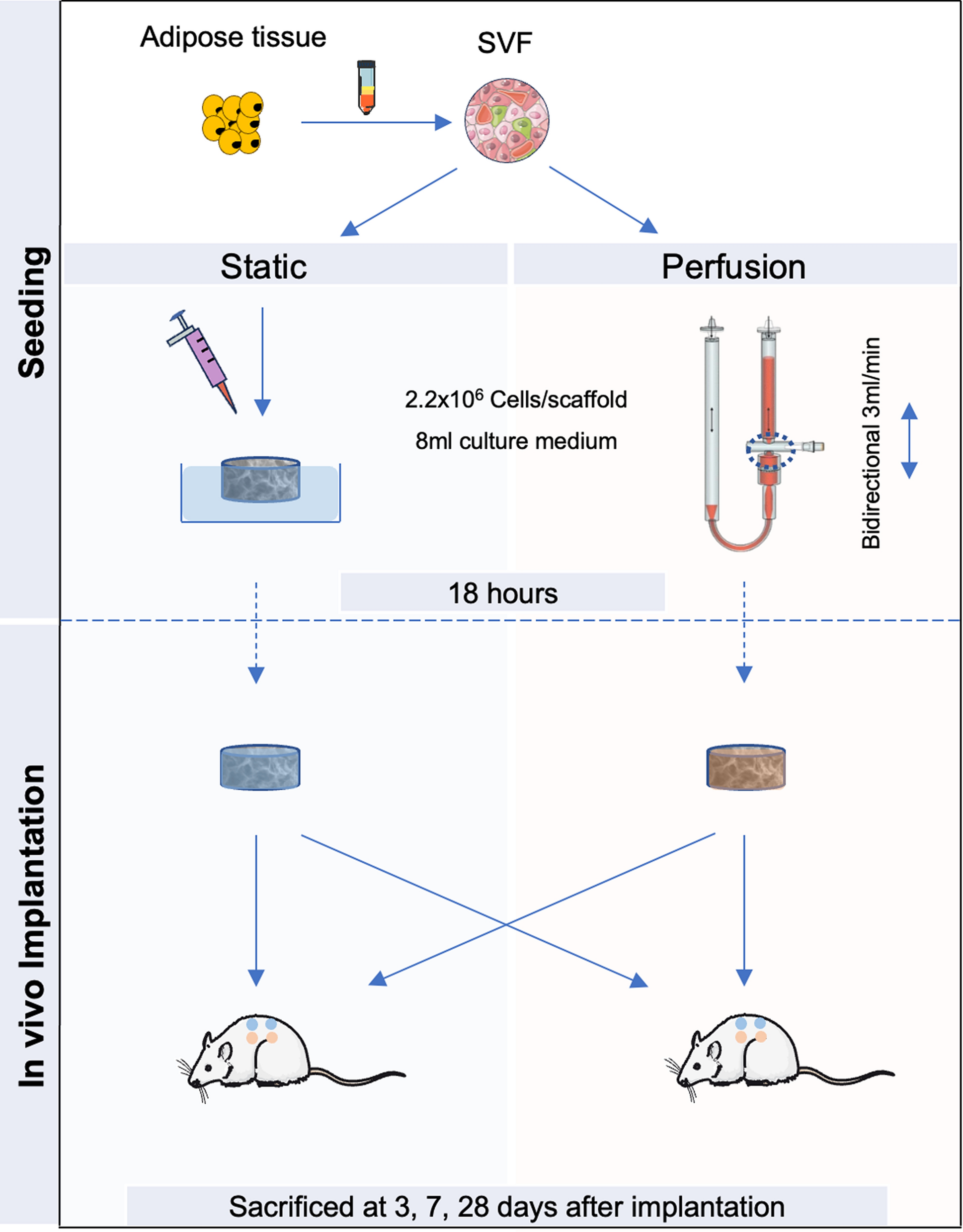
HFP-MSCs were seeded into either standard cell culture flasks or into vertical bioreactor culture chambers (PBSMini, PBSBiotech) after detachment from 90% confluent 2D cultures via accutase (GIBCO, #A1110501) and counting via trypan blue dye exclusion using a Neubauer counting slide. For 2D and 3D EV production protocols as described below and outlined in Fig. 1, HFP-MSCs were used having a passage number lower than 5 at seeding for EV production.
2D protocol
Cells were seeded at 10,000 per cm2 in T175 flasks in 30 ml growth medium and cultured until 80–90% confluency with growth medium change twice per week. The expansion phase usually lasted 7 days. Afterwards, medium was switched to serum free medium supplemented with 5% (v/v) of either FCS, CPRP or hypACT and 2 U/ml heparin (Gilvasan, #3,909,981) to mimic an intraarticular blood product treatment for 24 h. Then, medium was switched to 15 ml serum free medium to start EV collection phase. Conditioned media were harvested daily for 3 days and replaced with 15 ml fresh serum free medium. Harvested media were pooled per condition and donor and stored at -80 °C until further processing.
3D protocol
On average, 4.33*106 cells were mixed with 1.6 g Synthemax II microcarrier beads (Corning, #3781). The beads have a surface of 360 cm2/g, therefore the seed density corresponded to around 7,500 cells/cm2. Cells and microcarriers were suspended in 25 ml growth medium and put on a roller for 1 h at 37 °C with 5 rpm, with 2 min agitation phases and 5 min settling phases to allow for cell attachment. Then, the suspension was transferred to the PBSMini vertical wheel bioreactor chamber (PBSBiotech, USA) with a 25 ml pipette and 50 ml growth medium was added. Rotator speed was set to just keep the beads suspended. With increasing culture time, the wheel speed was increased from 18 to 25 rpm. During expansion phase, 20 ml growth medium was changed daily. Afterwards, medium was switched to serum free medium supplemented with 5% (v/v) of either FCS, CPRP or hypACT and 2 U/ml heparin (Gilvasan, #3,909,981) to mimic an intraarticular blood product treatment for 24 h. Then, the cell-bead suspension was transferred to 50 ml tubes for washing once with PBS via centrifugation at 200 x g for 5 min, before resuspending in serum free medium, returning to the bioreactor chamber and continuing culture in 80 ml total medium volume for 72 h for EV collection. Afterwards, conditioned supernatant was harvested via gravitational settling of the cell-microcarrier suspension and transfer of the clear supernatant to 50 ml tubes. After centrifugation at 4,000 x g for 15 min to remove residual cells and cell debris, the cleared conditioned supernatant was stored at -80 °C until further processing.
Ultrafiltration of conditioned supernatant
Conditioned supernatant stored at -80 °C was thawed at 37 °C in a water bath. Volumes of 45 ml or 80 ml were processed for 2D or 3D cultures, respectively. To prepare the 100 kD Amicon ultrafiltration units (Merck, #UFC9100), one spin with PBS followed by one spin of 70% ethanol, again PBS and one spin of ultrafiltrated 1% BSA in PBS was performed to ensure sterility and to reduce unspecific attachment of EVs to the filter unit during sample processing. Conditioned supernatants were processed in aliquots of 15 ml per centrifugation round (4,000 x g, 15 min, 4 °C) using the same filter unit per sample to accumulate retentates. After the whole conditioned supernatant was filtered and EVs accumulated in the filter unit, 15 ml PBS were added to the filter unit to wash the EVs via resuspending the retentate and centrifuging again at 4,000 x g for 15 min at 4 °C. The primary retentate was then recovered as fraction “EV” via pipetting into 1.5 ml low attachment tubes (Biozym, #710,176). Then, 1 ml PBS was added to the filter unit, which was then vortexed at 2,000 rpm for 20 s to detach remaining EVs from the filter membrane. This cleanout procedure was performed 5 times consecutively to obtain fractions “W1”-“W5” to achieve quantitative recovery of EVs.
Nanoparticle tracking analysis (NTA)
Size and concentration of particles were determined via NTA in scatter mode on a Particlemetrix MONO device, while colocalisation experiments were performed on a TWIN device. Samples were diluted as required 1:100-1:1000 with PBS. Firstly, scatter mode video acquisition at 11 positions with 30 frames per second for 2 s each used the camera settings sensitivity 80 and shutter 100. Video acquisition and analysis was done with ZetaView 8.04.02 or Zetanavigator 1.0.3.8.6 and ParticleExplorer 4.0.5.4 software. Secondly, for colocalisation of CD9 and CD73 on EV samples, antibodies were 1:10 prediluted in PBS. 8 µl of EV suspension were mixed with 1 µl CD9 1:10 and CD73 1:10 and incubated in the dark for at least 1 h. Afterwards, stained EV suspensions were diluted 1:333 in PBS for measurement at camera sensitivity 95 to determine the percentage of CD9 positive particles colocalising with CD73 and vice versa.
Induction of senescence
To prepare control protein extracts of HFP-MSCs to rule out senescence induction due to bioreactor culture, HFP-MSCs were seeded at 10,000 per cm2 in 6-well plates in growth medium. After 48 h, 5 µM etoposide (Sigma, # e1383) were added for 48 h as previously described [16], or 30 µM H2O2 (approximately 0.001% (w/v) for 2 h were added similar to a previous study [10]. Afterwards, cells were processed for protein sample preparation as described below.
Protein quantification
Total protein was determined via BioRad DC assay in microplate format. Ten microlitres of undiluted or diluted sample as required were used. Samples were lysed with 10 µl RIPA buffer without protease inhibitors (Thermo Scientific, #89,900) for 10 min at 4 °C to release EV protein content prior to performing the assay. A linear standard curve ranging from 2.0 to 0 mg/ml was generated with bovine serum albumin (BSA) (Sigma-Aldrich, #A8022). After 15 min incubation in the dark, absorbance at 670 nm was determined using a plate reader.
Western blot
HFP-MSCs or HFP-MSC EVs were lysed in RIPA buffer (Sigma #R0278) supplemented with phosphatase and protease inhibitor cocktail (Thermo Scientific, #78,440) via vigorous pipetting and incubation at 4 °C for 15 min. Following protein quantification, 10 µg total protein were separated on 10% or 4–12% gradient SDS-PAGE pre-cast gel electrophoresis (Invitrogen, #NP0322). Samples were reduced via 2 µl 1 M dithiothreitol for detection of Alix, ApoB100, p21 and (cleaved) caspase 3, while samples were separated unreduced to probe for CD9 and CD63. After semidry blotting onto PVDF membranes, the following primary antibodies were used 1:1000 diluted in 1% BSA/PBST: Alix (Cell Signaling, #2171), CD63 (BioLegend, #353,005), CD9 (System Biosciences, #EXOAB-CD9A-1), ApoB100 (SantaCruz, #sc-393,636), caspase 3 (Cell Signaling, #14,220), cleaved caspase 3 (Cell Signaling, #9661), p21 (Cell Signaling, #2947), GAPDH (Abcam, #ab37168). Detection was performed via enhanced chemiluminscence (ECL) using WesternBright ECL substrate (Advansta, #K-12,045-D20). Automatic white correction was applied to blot images via GIMP software version 2.10 before semiquantitative analysis was performed densitometrically using the GelAnalyzer plugin of ImageJ software version 1.51s.
RNA extraction and reverse transcription quantitative PCR (RT-qPCR)
Total RNA was extracted from chondrocytes cultured in the inflammation model and M1 macrophages using a High Pure RNA Isolation Kit (Roche, #11,828,665,001). A Transcriptor cDNA Synth Kit (Roche, #04897030001) was used to synthesize cDNA according to the manufacturer’s protocol. For qPCR, FastStart Essential DNA Probes Master (Roche, #06402682001) was mixed with 1 µL cDNA and 900 nM of primer (Supplementary table 1). CT values were obtained on a LightCycler 96 device (Roche, #05815916001). Data were normalized to GAPDH, and fold changes were calculated via the ΔΔCt method.
Statistics
Statistical analyses were performed via GraphPad Prism 9.5 and statistical significance was accepted for p < 0.05. Data were tested for normality via Shapiro-Wilk test and analysed via one-way ANOVA with Tukey post-hoc test or two-way ANOVA with Sidak post-hoc test as indicated in the figure legends. Correlation analyses were performed via Pearson or Spearman correlation analysis for normally or not normally distributed data, respectively. Data in graphs are represented as mean ± SD, unless otherwise stated.







Comments (0)