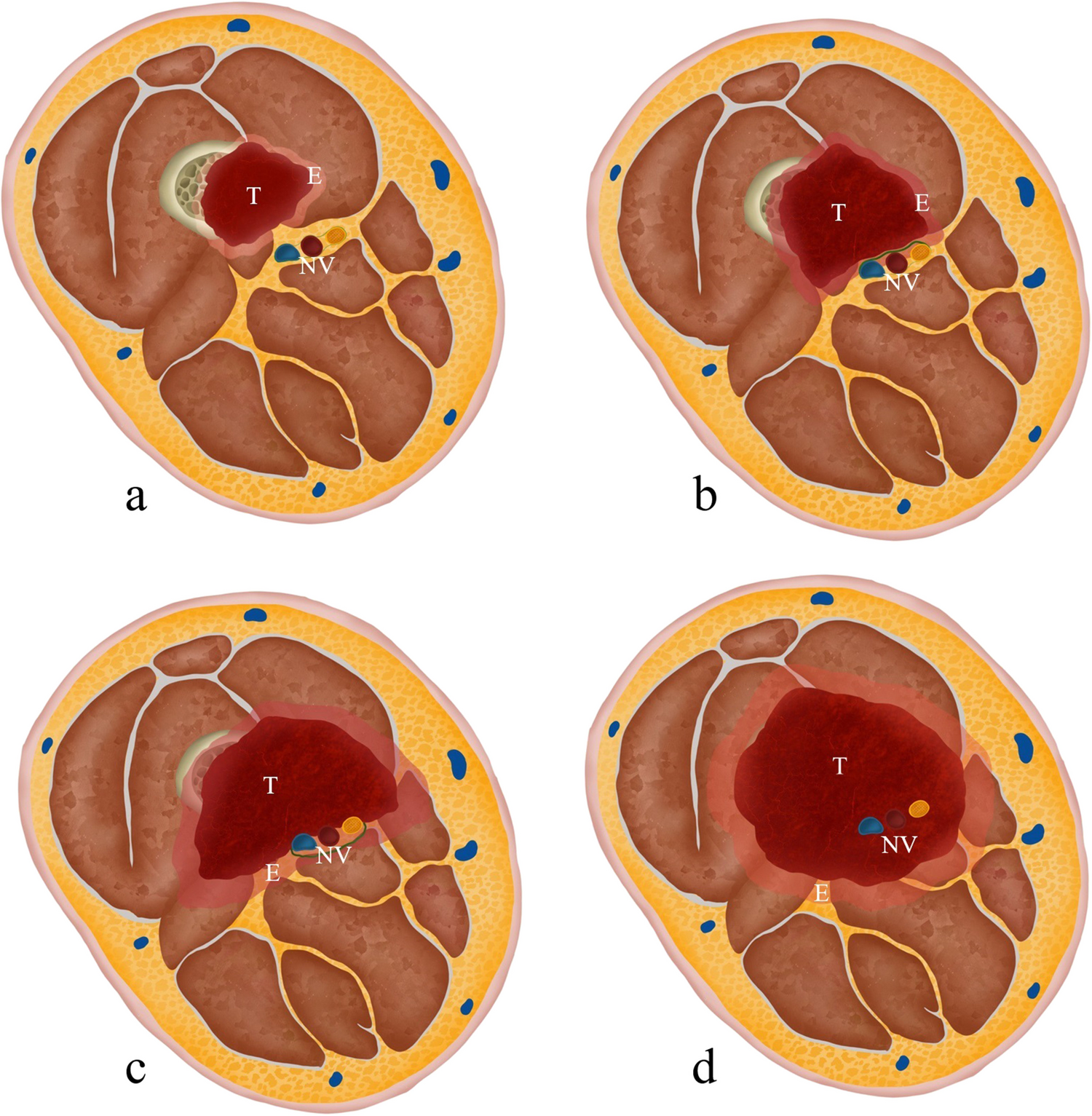
We report metachronous osteoclast-rich tumours occurring in a patient with polyostotic PDB. The initial clinical and imaging concerns were of PDB-related malignancy or cGCTB, and metastasis was considered when a mass with similar imaging appearances developed in the cervical spine four months after the iliac lesion was treated. Although both lesions were histologically benign, the pelvic lesion did not exhibit the typical morphological features or the hallmark genetic alteration, a mutation in H3F3A/B G34, of cGCTB. Furthermore, the cervical lesion shared morphological similarities with cGCTB, but, similar to the pelvic lesion, did not harbour an H3F3A/B G34 genetic alteration. Subtle genetic differences between the two lesions showed that the cervical lesion was not a metastasis from the pelvic tumour. Our analysis also confirmed that these osteoclast-rich tumours were genetically distinct from cGCTB.
The possibility exists that these OC-rich lesions represent an extreme manifestation of PDB. A “true” neoplasm would be expected to harbour a somatic driver genetic alteration, which could not be identified in any of the samples analysed. As such, these masses were more consistent with “tumour-like” lesions. The cause of their large size is unclear, but abnormal paracrine stimuli have been shown to play a key role in the initiation and growth of cGCTB, as recently reported by Cottone et al. [5], and represents a potential contributing factor.
From a therapeutic perspective, the clinical management of cGCT in long bones is curettage or excision. However, when a lesion is inoperable, or where resection would be associated with major morbidity, particularly in the pelvis and spine, downstaging prior to surgery with bisphosphonates, RANK ligand inhibitors, tumour embolisation or radiation therapy may be employed [6,7,8]. Clinical management of osteoclast-rich lesions in PDB is less well defined, reflecting their rarity. If a large symptomatic tumour is present, as in our case, surgical removal is likely to be advocated. Antiresorptive agents, which are known to control Paget bone disease [9] through inhibition of osteoclast formation/activity, may provide a valuable option for the treatment of small lesions. To our knowledge, the efficacy of this treatment in Pagetic osteoclast-rich lesions has not been evaluated and would be difficult to prove because the tumours are so rare.
From a diagnostic perspective, the finding of an osteoclast-rich lesion without H3F3A mutations should prompt further investigations and PBD should be considered. A diagnosis of PBD has implications for patients and their families, and detection of a germline alteration would allow screening of family members, provision of an early diagnosis and non-invasive monitoring of those harbouring the mutation. Furthermore, antiresorptive agents, such as bisphosphonates and denosumab, already used in the treatment of PBD without osteoclast-rich lesions [9], could potentially avoid the development and progression of such lesions in individuals with germline alterations in ZNF687 and PFN1. Importantly, screening for genetic drivers in PBD is valuable as there remains a significant proportion of patients in whom the genetic basis of the disease is not yet explained [1].




Comments (0)