
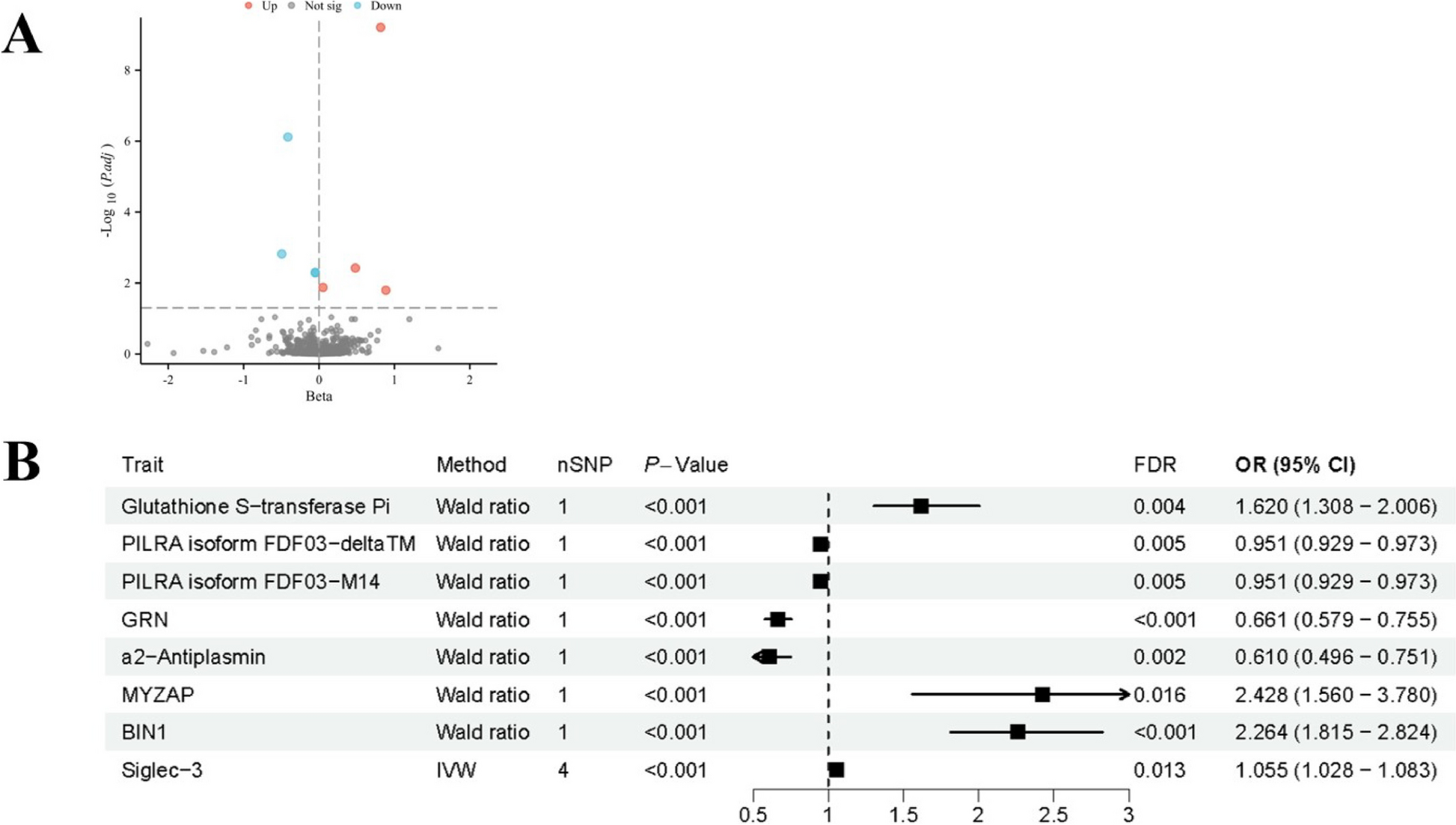
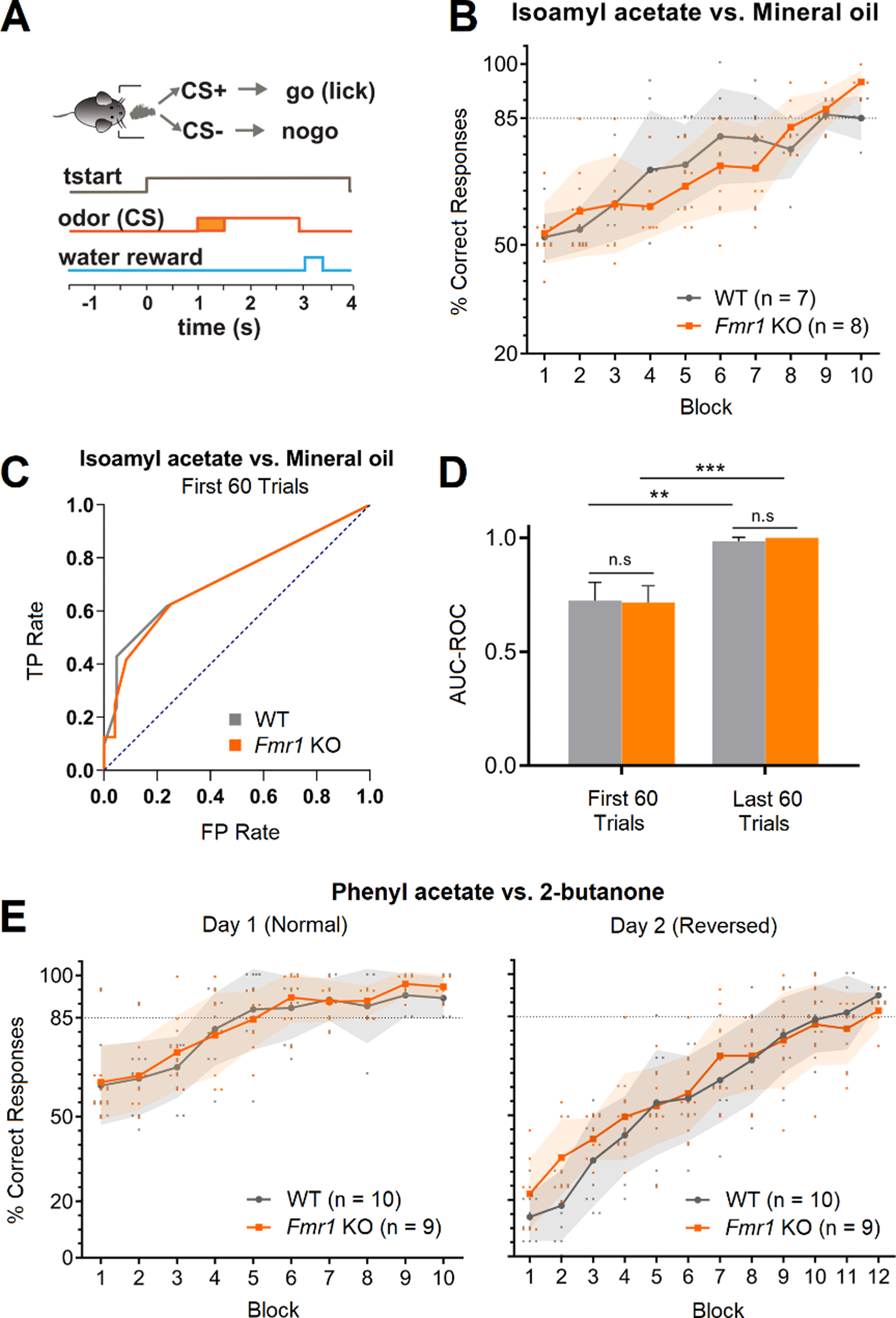
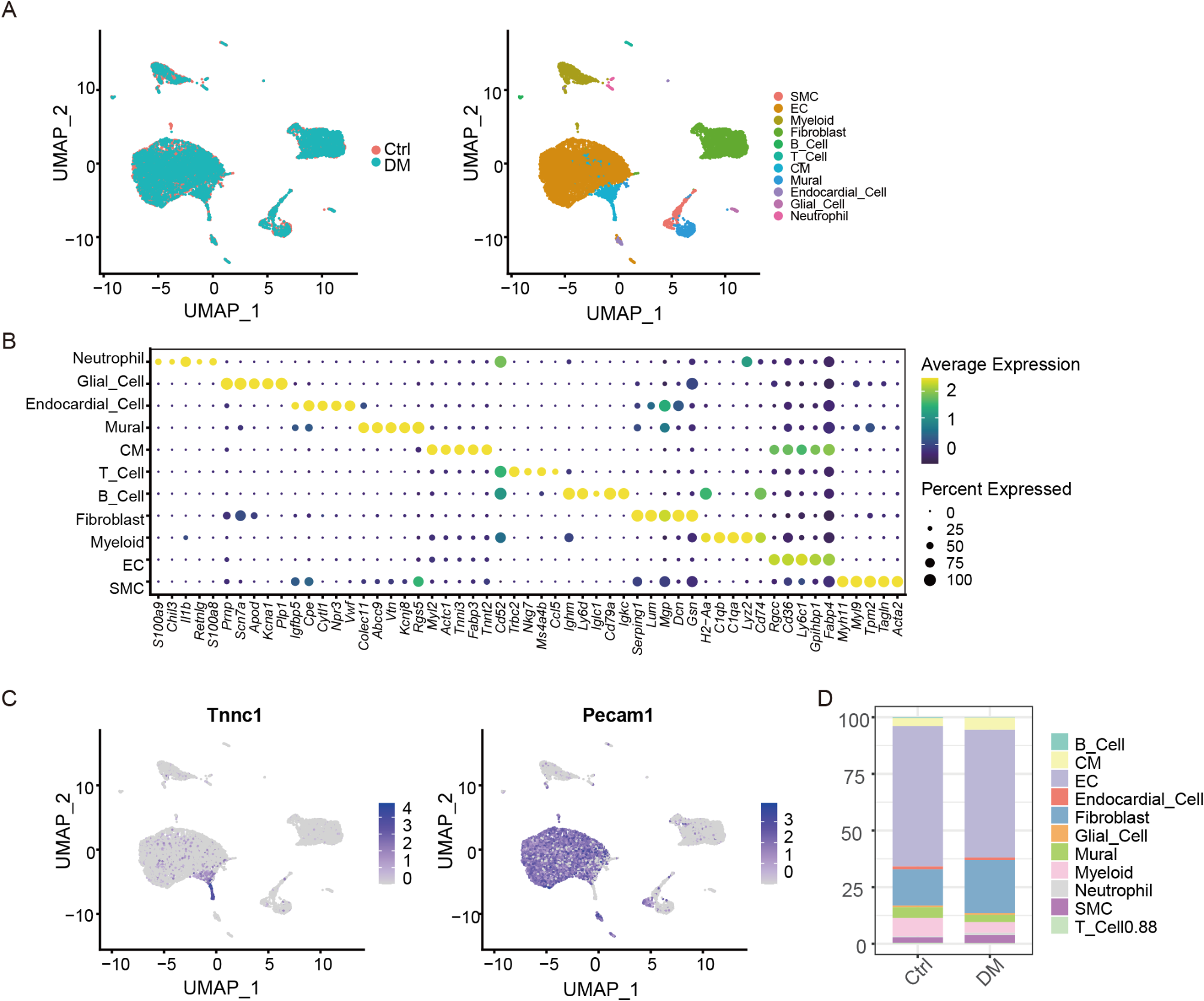
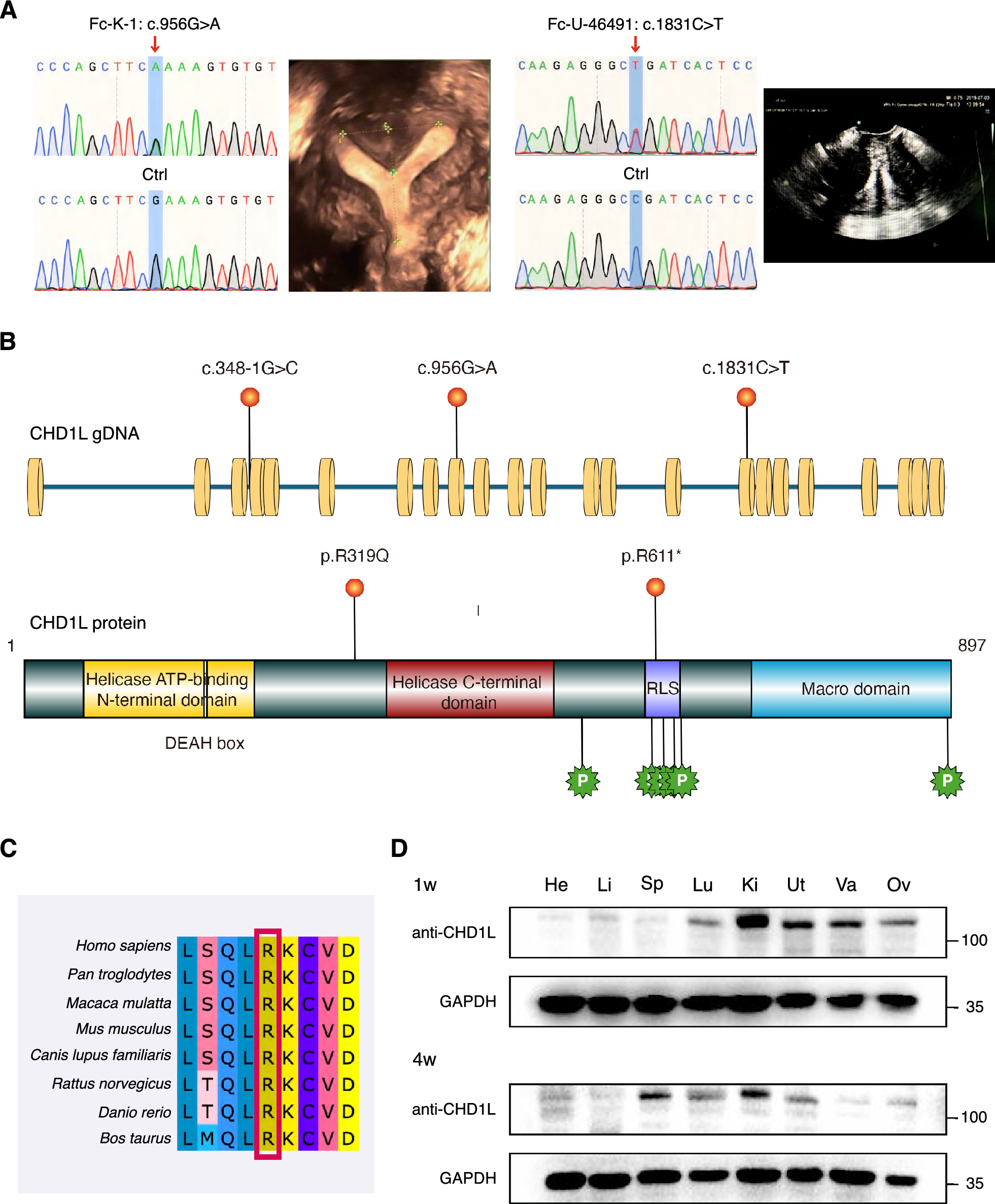
Remember me






The essential role of FGF-FGFR1 signaling in NPCs is well-documented [29]. Previous research has also demonstrated the development of a bivalent FGFR1 agonist to maintain and support the pluripotency of human embryonic stem cells (HESCs) [30]. Similar strategies for developing DNA-based agonists have been previously employed to mimic HGF and VEGF [31, 32]. However, the effort to use DNA-based receptor agonists for NPC stemness maintenance remains unexplored. In this study, we intended to utilize a DNA-based synthetic agonist to mimic the function of bFGF, allowing for controlled receptor dimerization and effective activation of FGFR signaling in NPCs. Similar to the previous concept, we designed a bivalent DNA architecture containing two FGFR1-binding domains that could facilitate FGFR1 dimerization. We employed a previously characterized monomeric FGFR-binding aptamer, a 38-mer stem-loop oligonucleotide, as our FGFR1 binder (Fig. 1A, Table S1). To induce ligand-mediated receptor dimerization, we conjugated two monomeric FGFR1 binders to create a bivalent ligand capable of assembling two cell surface FGFR1 molecules, acting as an FGFR1 agonist. Structural predictions indicated that the bivalent configuration did not adversely affect the secondary structure of each FGFR1-binding domain (Fig. 1A). Using 8% Native PAGE gel electrophoresis, we observed that the molecular weight of a single-stranded FGFR binder was approximately 25 nucleotides (nt). In contrast, the molecular weights of both the bivalent FGFR1 agonist and its control oligonucleotide were around 72 nt, likely due to their respective secondary structures (Fig. 1B). Notably, the control oligonucleotide (Ctrl-oligo), which has same number of nucleotides with a scrambled sequence compared to the FGFR1 agonist, displayed a higher molecular weight during electrophoretic analysis. This observation may be attributed to the G-quadruplex (G4) structure possessed by the FGFR1 agonist, which leads to a more compact conformation in native PAGE assays [30]. In contrast, the scrambled sequence in the control group disrupts this ordered structure, resulting in a more linear spatial configuration. Consequently, the electrophoretic migration rate slows down, manifesting as a higher molecular weight. This data also suggests that the compromised structure of the Ctrl-oligo could adversely affect its ability to effectively interact with FGFR1.
Fig. 1
Design and characterization of FGFR-agonist. (A) The secondary structures of the FGFR-binder and FGFR-agonist determined using Mfold software are presented, indicating the formation of a stem-loop structure. (B) The secondary structure of DNA-based FGFR-agonist is verified using native polyacrylamide gel electrophoresis (PAGE). This panel also includes the determined molecular weights of the single-stranded FGFR-binder, FGFR1-agonist, and a control oligonucleotide (Ctrl oligo). (C) SPR sensorgrams showing the real-time binding kinetics of FGFR-agonist aptamer to immobilized FGFR1 extracellular domain at various concentrations. The sensorgrams are color-coded based on the concentrations of the FGFR-binder (1, 2, 4, 8, 16 and 32 nM) and FGFR-agonist (0.16, 0.31, 0.625, 1.25, 2.5, 5 and 10 nM), respectively. (D) The relative binding performance of FGFR-binder and FGFR-agonist to the NIH3T3 cells was determined by flow cytometry (refer to Fig. S1 for additional details)
To elucidate the molecular binding properties of the monomeric FGFR-binder and bivalent FGFR1-agonist with the extracellular domain of FGFR1, we employed Surface Plasmon Resonance (SPR) analysis using the Biacore method. Our results revealed that both FGFR-binder and FGFR1-agonist exhibited high binding activity against FGFR1 protein (Fig. 1C). However, there are distinct differences in the binding kinetics and affinities between the two constructs (Fig. 1C and Table S2). The FGFR-binder displayed a dissociation constant (KD) of 2.5 nM, while the FGFR1-agonist demonstrated a substantially improved affinity, with a KD value reaching 46.1 pM. The binding kinetics analysis revealed that the FGFR-binder had a fast association rate (Ka) of 2.20 × 1010 Ms− 1 and a dissociation rate (Kd) of 54.9 s− 1, indicating a rapid off-rate (Table S2). In contrast, the FGFR-agonist had a Ka of 4.51 × 106 Ms− 1 and a Kd of 2.08 × 10− 4 s− 1. The dissociation speed of the FGFR1-agonist was significantly slower than that of the FGFR-binder, implying a more stable binding interaction between FGFR-agonist and FGFR1 protein, probably due to the multivalent interactions facilitated by the bivalent FGFR-agonist.
We next examined the binding activity of FGFR-agonist to the receptors on living cells using flow cytometry. We employed an FGFR1-positive NIH3T3 cell line, a murine fibroblast cell line. The results demonstrated that the FGFR1-binder exhibited a high affinity for FGFR1 with a KD of 39 nM (Fig. 1D and S1). Notably, the bivalent FGFR1-agonist could affect the cell-binding affinity compared with the monomeric FGFR-binder while significantly increasing the binding fraction, consistent with the previous finding in vitro SPR experiment. To explore the functionality of FGFR-agonist on the FGFR activation, the serum-starved NIH3T3 were stimulated with the FGFR-agonist and bFGF for 10 min, and the phosphorylation of FGFR1 (Tyr653/654) was evaluated using an enzyme-linked immunosorbent assay (ELISA). The results showed that the FGFR-agonist significantly promoted the phosphorylation level of FGFR1 compared with the controls treated with the monomeric FGFR-binder (Fig. S2). Moreover, the stability of the FGFR-agonist under physiological conditions would be highly important for its robust effect for in vivo application. To characterize this feature, we performed a serum stability assay by incubating the FGFR-agonist with 10% FBS at 37 °C. After incubation for different durations, we analyzed all samples using 8% Native PAGE gel electrophoresis. The results showed that the FGFR-agonist remained stable after 16 h in 10% FBS, indicating its suitability for subsequent experiments (Fig. S3).
FGFR-agonist promotes FGFR signaling in NPCsHaven successfully constructed a serum-stable DNA-based FGFR-agonist with high binding affinity for FGFR1, we next explored the potential FGFR-agonist in modulating the FGFR signaling pathway of NPCs. We obtained HESC-derived NPCs using a previously described pipeline with different combinations of culturing medium (Fig. S4). Immunofluorescence analysis confirmed the positive staining of these cells with Nestin and Pax 6, both of which are NPC markers (Fig. S5). Upon neuronal differentiation without bFGF, the NPCs were able to differentiate into MAP2-positive neurons (Fig. S6).
To evaluate the effectiveness of the FGFR-agonist in activating FGFR1, we employed an ELISA to quantify the phosphorylation levels of FGFR1 (Tyr653/654) in NPCs. Following a 24-hour period of serum starvation to synchronize cellular responsiveness, NPCs were exposed for 10 min to either bFGF or our custom-synthesized FGFR-agonist. Notably, the FGFR-agonist treatment resulted in a significant 1.4-fold increase in FGFR1 phosphorylation compared to the control group treated with Ctrl-oligo (Fig. 2A). Although the phosphorylation levels elicited by bFGF were higher (2.4-fold), our findings indicate that FGFR-agonist effectively mimics the activation properties of the natural ligand, bFGF. In parallel, we explored the activation of downstream signaling pathways, focusing on ERK1/2 phosphorylation at residues Thr202/Tyr204. Western blot analyses confirmed a pronounced increase in ERK1/2 phosphorylation levels for NPCs treated with either FGFR-agonist or bFGF when compared to both untreated controls and Ctrl-oligo treated cells (Fig. 2B and S7). Quantitatively, the ERK1/2 phosphorylation levels were amplified 10.5-fold in the FGFR-agonist group relative to the Ctrl-oligo group, whereas the bFGF-treated group showed a 9.4-fold increase relative to the control group (Fig. 2C).
Fig. 2
FGFR-agonist promotes FGFR signaling of NPCs. (A) ELISA-based quantification of FGFR1 phosphorylation in NPCs. FGFR1 (Tyr653/654) phosphorylation levels were measured in serum-starved NPCs after 10-minute exposure to either bFGF (20 ng/mL) or FGFR-agonist (40 nM). Data are represented as mean ± SD (n = 3); **p < 0.01(unpaired two-tailed Student’s t-test). (B) ERK1/2 phosphorylation profile via Western Blot. Serum-starved NPCs were treated with either bFGF (20 ng/mL) or FGFR-agonist (40 nM) for 10 min. Phosphorylation of ERK1/2 (Thr202/Tyr204) was then examined by Western blot, with GAPDH serving as an internal control. (C) Quantification of relative ERK1/2 phosphorylation to total ERK. The phosphorylation levels of ERK1/2 were quantified and normalized to the control group without any treatment. Data are expressed as means ± SD (n = 3); **p < 0.01(unpaired two-tailed Student’s t-test). (D) Time-dependent Activation of ERK1/2. Serum-starved NPCs were exposed to either bFGF (20 ng/mL) or FGFR-agonist (40 nM) over varying time intervals (5, 10, 15, 30, 60 min). The phosphorylation of ERK1/2 (Thr202/Tyr204) was assessed via Western blot. (E) Quantification of the time-dependent data from panel D, illustrating the kinetics of ERK1/2 phosphorylation in NPCs when stimulated by either bFGF or FGFR-agonist. Data are presented as mean ± SD (n = 3); no significance (n.s.) was found, as determined by one-way ANOVA
To understand the temporal characteristics of FGFR-agonist-mediated signaling, we treated serum-starved NPCs with either bFGF or FGFR-agonist across multiple time intervals. The kinetics of ERK1/2 phosphorylation in response to FGFR-agonist were found to parallel those induced by bFGF (Fig. 2D). This corroborates that FGFR-agonist is comparably effective as bFGF in sustaining FGFR/ERK signaling over time (Fig. 2E). We also validated these findings across different cell lines by employing ATDC5 cells, a murine chondrocyte line that is responsive to bFGF stimulation [33]. We observed similar trends in FGFR1 and ERK1/2 phosphorylation, confirming the broad efficacy of the FGFR-agonist. Interestingly, bFGF elicited more sustained ERK phosphorylation in ATDC5 cells than FGFR-agonist, which differs from that shown in NPCs (Fig. S9). This variation could be attributed to differences in FGFR1 endocytosis across different cell types, which may consequently alter the kinetics of downstream signaling.
Collectively, our findings establish the role of the FGFR-agonist as an effective modulator of FGFR/ERK signaling, underscoring its ability to precisely modulate cellular signaling pathways and illuminate its potential as a potent tool for manipulating cellular behavior in NPCs and perhaps other FGFR-positive cell types.
FGFR-agonist recapitulates the function of bFGF to modulate NPCsBuilding on our previous findings concerning the role of the FGFR/ERK signaling pathway in regulating essential cellular functions, we further examined the effect of FGFR-agonist on the proliferation of NPCs. Employing the CCK8 assay, we found that FGFR-agonist markedly promoted NPC proliferation during both 24-hour and 72-hour treatment periods, achieving levels similar to those observed with bFGF treatment (Fig. 3A). Remarkably, a 72-hour treatment with FGFR-agonist led to a 4.4-fold increase in cell proliferation when compared to the control group treated with Ctrl-oligo. This result closely paralleled the 4.6-fold increase seen in the bFGF-treated group. We further assessed the directional migratory capabilities of NPCs upon stimulation with bFGF and FGFR-agonist employing a Transwell migration assay. The results demonstrated that FGFR-agonist significantly elevated the number of migrated NPCs compared to the control group treated with Ctrl-oligo (Fig. 3B and S10). Quantitative analysis revealed that FGFR-agonist increased the cell migration rate by 2.8-fold relative to the Ctrl-oligo group. In contrast, recombinant protein bFGF exhibited a stronger ability to induce directional migration, achieving a 5.6-fold increase compared to the control group (Fig. 3C). Given the previously observed similarity in the proliferative effects between FGFR-agonist and bFGF, these data suggest that the differential migration responses might be due to biased signaling activation, warranting further investigation into the underlying mechanisms. In addition, the function of FGFR-agonist was extended to the ATDC5 cells, where FGFR-agonist similarly promoted proliferation and enhanced wound closure rates, comparable to the effects of bFGF (Fig. S9). Therefore, our findings underscore the potential of FGFR-agonist as an effective alternative or adjunct to bFGF for stimulating both NPC proliferation and migration.
Fig. 3
FGFR-agonist regulates cellular behaviors of NPCs. (A) The proliferation of NPCs was evaluated at 24-hour and 72-hour time points following treatments with bFGF or FGFR-agonist, using a CCK8 kit. (B) Serum-starved NPCs were placed in the upper chamber, while bFGF(20 ng/mL) and FGFR-agonist (40 nM) were included in the lower chamber for 24 h and stained, followed visualization under light microscopy. Scale bar = 100 μm. (C) The data from the Transwell assay are quantified to assess the impact of different treatments on NPC migration. Data are presented as mean ± SD (n = 6). (D) Representative images showing the undifferentiated NPCs (Nestin-positive) and differentiated neurons (Tuj-1 positive) NPCs treated with bFGF, FGFR-agonist and Ctrl-oligo for 7 days and subjected to immunofluorescence staining. Scale bar = 100 μm. (E) The ratios of Nestin-positive cells and Tuj-1-positive cells are quantified against total cells (DAPI-positive). Data are presented as mean ± SD (n = 3), with **p < 0.001, as determined by unpaired t-tests. (F) Representative images of neurospheres generated by NPCs cultured in suspension for 10 days in the presence or absence of bFGF (20 ng/mL), FGFR-agonist (40 nM), or FGFR-agonist (200 nM). The scale bar = 100 μm. (G) Quantification of the changes in neurosphere volume over a 10-day culture period under various treatment conditions. Data are displayed as mean ± SD (n = 3), with **p < 0.0001, as determined by one-way ANOVA
Given the critical role of FGFR signaling in maintaining the stemness of NPCs, as indicated in previous research [18], we further explored the ability of FGFR-agonist to sustain the undifferentiated state of NPC. For this purpose, NPCs were treated with bFGF, FGFR-agonist, or Ctrl-oligo for 7 days. Immunofluorescence assays were performed to assess the expression levels of the stem cell marker Nestin and the neuronal differentiation marker Tuj-1. Our observations revealed that NPCs treated with either FGFR-agonist or bFGF retained a progenitor-like morphology and exhibited robust Nestin expression while showing minimal Tuj-1 levels (Fig. 3D and Fig. S11). In contrast, untreated cells or those treated with Ctrl-oligo displayed an elongated, neuron-like morphology and included some apoptotic cells, suggesting spontaneous differentiation (Fig. 3D). Quantitative analysis further substantiated these findings. The treatment with 40 nM of FGFR-agonist led to approximately 70% of cells being Nestin-positive and only 3.2% being Tuj-1-positive neurons (Fig. 3E). Treatment with 200 nM of FGFR-agonist resulted in an even higher proportion of Nestin-positive NPCs (91%) with a low ratio of neurons expressing Tuj-1 (1.3%). For comparison, bFGF treatment yielded 86% Nestin-positive cells and 2.5% Tuj-1-positive neurons (Fig. 3E). Collectively, our data strongly suggest that FGFR-agonist, much like bFGF, is effective in maintaining the undifferentiated state of NPCs and limiting spontaneous neuronal differentiation.
We next sought to investigate its potential utility in facilitating neurosphere formation, an essential procedure for obtaining neural progenitor cells for therapeutic applications. To this end, we compared its effects with bFGF, a standard growth factor ubiquitously used in NPC cultures. NPCs were cultured in a suspension system for a 10-day period, and two concentrations of FGFR-agonist (40 nM and 200 nM) were evaluated alongside a standard dose of bFGF (20 ng/mL). We evaluated several key parameters, including the number, size, and morphology of the neurospheres, to gauge the effectiveness and quality of neurosphere formation. Our preliminary findings showed that FGFR-agonist had a favorable impact on both the size and quantity of the neurospheres, akin to the effects observed with bFGF (Fig. 3F). In the presence of FGFR-agonist, neurospheres displayed morphological integrity comparable to, or even better than, those maintained with bFGF throughout the 10-day culture period (Fig. 3F). Conversely, the untreated control group merely formed rudimentary cell clusters within the initial five days. Subsequent quantitative analyses revealed that the dimensions of the neurospheres in the FGFR-agonist-treated groups were on par with those in the bFGF-treated group (Fig. 3G). By day 3, a clear divergence was evident in the growth trajectories between the neurospheres treated with bFGF or FGFR-agonist (40 nM) and those in the untreated group. This distinction persisted until day 10 when neurospheres in the active treatment groups continued to expand while those in the control group began to disintegrate. Furthermore, our 7-day treatment study revealed stable maintenance of neurosphere circularity in both bFGF and FGFR-agonist treatment groups. Specifically, treated neurospheres exhibited significantly higher circularity levels than untreated cells or those treated with Ctrl-oligo (Fig. S12). Collectively, these results validate the utility of FGFR-agonist as an effective substitute or supplement for bFGF in the formation and maintenance of neurospheres, providing compelling evidence that FGFR-agonist can emulate the biological roles of bFGF in regulating NPC behavior and promoting neurosphere development.
FGFR-agonist affects the transcriptome program of NPCsTo explore the effects of the FGFR-agonist on NPCs at the transcriptomic level, we performed nanopore sequencing and conducted bioinformatic analysis on the neurospheres maintained with the FGFR-agonist and bFGF for 10 days. Total RNA was extracted from the neurospheres, and nanopore sequencing was employed to obtain long-read sequencing data capable of detecting full-length transcripts. The sequencing data were processed and analyzed to identify differentially expressed genes (DEGs) and investigate their functional significance. We first compared the transcription profiles of NPCs treated with the FGFR-agonist and bFGF, and the results showed that both upregulated and downregulated gene profiles were highly similar between the two conditions compared to the untreated NPCs (Fig. 4A). The similarity parameter of the transcription profiles between the FGFR-agonist and bFGF-treated NPCs was 0.996 (Fig. S13). Specifically, we identified 111 upregulated genes and 97 downregulated genes in NPCs treated with the FGFR-agonist compared to the untreated group (Table S3). The volcano plot of DEGs demonstrated the significant differential expression of transcripts between the FGFR-agonist-treated and the untreated groups (Fig. S14). Functional enrichment analysis of the upregulated DEGs revealed significant enrichment in gene ontology (GO) terms related to stem cell maintenance, including signaling pathways regulating pluripotency of stem cells, MAPK signaling pathway, Wnt signaling pathway, PI3K-Akt signaling pathway, and Hippo signaling pathway (Fig. 4B) [34]. Additionally, the upregulated DEGs showed significant enrichment in signaling pathways associated with NPC maintenance and self-renewal, such as forebrain development, regulation of ERK1 and ERK2 cascade, and cellular responses to platelet-derived growth factor stimulus (Fig. 4C). We focused on the KEGG pathway of regulating pluripotency of stem cells and found that several pivotal neural stem cell markers were upregulated in the FGFR-agonist treated group (Fig. S15). Notably, SOX2 and c-Myc, instrumental transcription factors in promoting self-renewal and pluripotency of embryonic or neural stem cells, were among the genes displaying upregulated expression following FGFR-agonist treatment [35, 36].
Fig. 4
FGFR-agonist programmed transcriptional profile of NPCs. (A) Comparative gene expression heatmaps in NPCs treated with FGFR-agonist and bFGF. The heatmaps display the transcriptional profiles of NPCs exposed to FGFR-agonist or bFGF for 10 days, relative to untreated NPCs (None). Both upregulated (red) and downregulated (green) gene profiles demonstrate a high degree of similarity between the two treatment conditions. (B) Functional enrichment analysis of upregulated DEGs in FGFR-agonist-treated NPCs versus untreated NPCs, utilizing gene ontology (GO) terms. It displays the top 10 significantly enriched GO terms, with the corresponding number of genes in parentheses. (C) A KEGG pathway enrichment analysis of the upregulated DEGs in FGFR-agonist-treated NPCs compared to untreated NPCs, indicating the top 20 significantly enriched pathways, denoted by the -log10 (adjusted p-value). (D) A similar KEGG pathway enrichment analysis for downregulated DEGs in FGFR-agonist-treated NPCs compared to untreated NPCs shows the top 20 significantly enriched pathways. (E) This panel presents a pathway enrichment analysis of differentially expressed transcripts in FGFR-agonist-treated NPCs versus untreated NPCs, based on KEGG pathway classification, highlighting the top 20 significantly enriched pathways. (F) A KEGG pathway enrichment cnet plot for upregulated DEGs in FGFR-agonist-treated NPCs compared to untreated NPCs. The nodes symbolize KEGG pathways, and the edges signify overlapping genes between pathways. Node size corresponds to the number of genes in the pathway, while node color represents the -log10 (adjusted p-value) of enrichment
Furthermore, the KEGG pathway enrichment analysis of the downregulated genes upon FGFR-agonist treatment revealed that the top five related neuronal diseases were associated with neuronal dysfunctions, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and Amyotrophic lateral sclerosis. This bioinformatic data suggests the potential of the FGFR-agonist to decouple mature neuronal function (Fig. 4D). These findings highlight the potential of the FGFR-agonist in promoting and preserving the stemness of neural progenitor cells. The pathway enrichment revealed that several important signaling pathways for NPCs were upregulated, including the Ras signaling pathway, Wnt signaling pathway, PI3K-AKT pathway, and cell cycle (Fig. 4E). The KEGG pathway enrichment plot analysis of the upregulated genes identified three signaling networks related to cell-substrate adhesion, ERK1/2 cascade, and response to stimulus (Fig. 4F). These findings indicate that the FGFR-agonist can effectively mimic the function of bFGF in maintaining the stemness of NPCs. Our results suggest that the FGFR-agonist can promote stem cell maintenance, regulate cell responses to growth factor stimuli, and suppress neuronal differentiation. These findings further support the potential of the FGFR-agonist as an effective modulator of NPCs, comparable to the role of bFGF.
FGFR-agonist maintains NPCs for long-term propagationNext, we investigated the applicability of the FGFR-agonist for long-term maintenance of NPCs. We successfully passaged and maintained the NPCs for over 50 passages using media containing the FGFR-agonist. Throughout the passages, we did not observe any significant changes in morphology or growth characteristics of the NPCs, indicating the stability and effectiveness of the FGFR-agonist in maintaining long-term self-renewal of NPCs (Fig. 5A). The absence of both bFGF and FGFR-agonist resulted in a decrease in cell survival over time, highlighting the essential role of the FGFR-agonist in promoting cell survival and self-renewal of NPCs. Furthermore, we explored the potency of the FGFR-agonist in maintaining NPCs for neuronal differentiation. We replaced the media of the FGFR-agonist-maintained NPCs with different media containing bFGF, FGFR-agonist, or a differentiation condition. After 10 days of incubation without bFGF or FGFR-agonist, the cells exhibited elongated morphology resembling neurons and the immunofluorescence analysis revealed a significant increase in Tuj-1-positive neurons and a decrease in Nestin positive NPCs in the NGF-mediated neuronal differentiation group compared to the bFGF or FGFR-agonist treated groups (Fig. 5B). This data indicates that in the absence of FGFR-agonist, the NPCs undergo neuronal differentiation. In contrast, the FGFR-agonist treated NPCs maintained a strong staining signal for Nestin, indicating the preservation of the undifferentiated state. Together, these findings validate the functionality of the FGFR-agonist in maintaining the multipotency of NPCs for neurogenesis. Therefore, our results demonstrate that the DNA-based FGFR-agonist can serve as an effective alternative to bFGF in maintaining the stemness of NPCs while decoupling neuronal differentiation. The use of artificial agonists would provide a valuable tool for large-scale acquisition of neural cells for functional assessment, disease modeling, and potential applications in regenerative medicine.
Fig. 5
The potency for neurogenesis of long-term propagated NPCs by FGFR-agonist. (A) The influence of FGFR-agonist on proliferative capacity of the NPCs after 50 passages. NPCs maintained in FGFR-agonist were propagated for 50 passages and treated with either bFGF, FGFR-agonist or Ctrl-oligo for 72 h. The cell proliferation was assessed using the CCK8 assay. Data are depicted as mean ± SD (n = 5), with *p < 0.001 (one-way ANOVA). (B) Long-term propagated NPCs underwent different treatment regimens, including bFGF, FGFR-agonist, or NGF-induced differentiation, over a 10-day period. Following treatment, cells were fixed and analyzed via immunofluorescence. Nestin is represented through green fluorescence, while Tuj-1 is indicated by red fluorescence. Cell nuclei are counterstained with DAPI for contrast. Scale bar = 100 μm. Specific regions of interest (ROI), delineated by white-bordered boxes, are further magnified to provide a detailed view, accompanied by a zoom-in scale bar set at 10 μm
Comments (0)