
Remember me
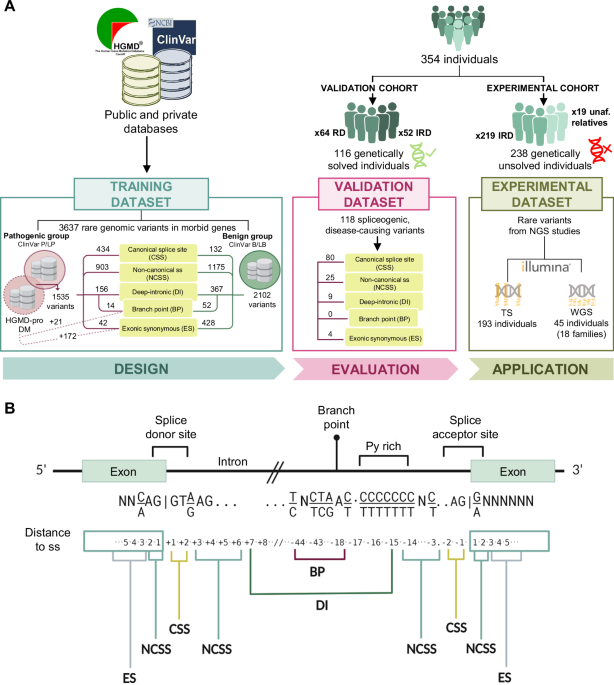

Glioblastoma (GBM) tissue, unlike that of low-grade gliomas (LGGs), has been shown to display extensive and heterogeneous ECM deposition and tissue remodeling23. Thus, we began our analysis with an unbiased characterization of the ECM transcriptome in GBM (Fig. 1a). First, we batch corrected (Supplementary Fig. 1a) publicly available GBM RNA-sequencing (RNA-seq) profiles from three independent datasets (TCGA, CGGA-693, CGGA-325) across 558 patients and performed non-negative matrix factorization (NMF) on the core matrisome gene expression matrix, thereby limiting our analysis to genes encoding structural components of ECM19. Based on cophenetic correlation coefficient (Supplementary Fig. 1f) and results from hierarchical clustering (Supplementary Fig. 1d, e), we grouped the data into two classes and defined a set of 505 samples as core samples based on a positive silhouette coefficient (Supplementary Figure 1b). Bootstrap resampling of the core matrisome gene set showed that clustering results were stable to variations in the gene set when compared to a reference gene set with a similar expression distribution (Methods, Supplementary Fig. 1c).
Fig. 1: Unsupervised analysis of retrospective RNA-seq data reveals two extracellular matrix states in glioblastoma that are prognostic of clinical outcome and conserved across multiple central nervous system cancers.
a Outline of the computational workflow. b ECMhi and ECMlo signature gene expression in adult (left) and pediatric (right) central nervous system tumors. Select signature genes are labeled. c Frequency of ECM states across different CNS tumors. d Kaplan–Meier survival curves for ECMhi (red) and ECMlo (blue) tumors of different histologies. Only tumors with a statistically significant effect (p < 0.05, log-rank test) are shown. e Gene ontology (GO) terms enriched in ECMhi (left) and ECMlo (right) states in adult gliomas. Top 10 GO terms are shown. f Functional gene expression signatures from Bagaev et al. (2021) in pediatric and adult brain tumors, grouped by ECM state.
In order to characterize the two classes/subtypes and classify external samples, we performed differential expression analysis on core samples between classes. For each class, we identified 49 signature genes as the intersection of top marker genes across the three datasets analyzed (Fig. 1b, Supplementary Data), such that differential expression of each signature gene was supported in all three datasets (Supplementary Fig. 1g). One cluster was characterized by upregulation of genes encoding fibrillar ECM proteins, including collagens (COL5A2, COL1A2, COL3A1, COL1A1, COL6A3, COL5A1, COL6A2, COL8A1) and glycoproteins (LAMB1, LAMC1, POSTN, FN1). All the signature genes encoding ECM proteins, with the exception of COL8A1 and LTBP2, have been shown to be present in GBM at the protein level (Supplementary Table 1). The clusters were defined as ECMhi, extracellular matrix high, (48.2% of samples) and ECMlo, extracellular matrix low, (51.8% of samples) to reflect their distinct ECM composition.
Next, to verify the existence of ECMhi and ECMlo subtypes in other CNS malignancies, we scored additional RNA-seq data spanning adult low-grade (n = 1164) and pediatric (n = 977) brain tumors as well as brain metastases (Bmets, n = 47) for expression of ECMhi and ECMlo signature genes. We found 83% of adult gliomas, 80% of pediatric gliomas, and 77% of Bmets could be reliably classified into either subtype, indicating that these programs are conserved across a range of CNS malignancies beyond GBM. The remaining samples were defined by contemporaneous up- or down-regulation of ECMhi and ECMlo signature genes and could not be reliably classified as either ECMhi or ECMlo. Principal component analysis (PCA) of core matrisome gene expression placed these samples between ECMhi and ECMlow samples, suggesting that they display an intermediate ECM state (ECMint) (Supplementary Fig. 1h). In adult gliomas, ECMhi subtype was associated with higher tumor grade, absence of IDH mutations, and mesenchymal subtype in GBM, whereas the majority of ECMlo GBM tumors were of the proneural subtype (Supplementary Fig. 1k) (P < 2.2E-16, Chi-Square Test). Pediatric tumors exhibited a highly skewed distribution of ECM states, where a number of tumors including craniopharyngioma (86%), neurofibroma (95%), meningioma (97%), and schwannoma (100%) were classified almost exclusively as ECMhi, whereas glioneuronal tumors (GNT, 3%) and medulloblastomas (4%) were classified almost exclusively as ECMlo (Fig. 1c), reflecting an association between the ECM subtypes and the intrinsic biology of different brain cancers. However, we did not detect an association between ECMhi score and the level of malignancy of pediatric brain tumors (Supplementary Fig. 1i), suggesting that the effect of ECM stroma may be context-specific. In astrocytoma and mixed glioma tumors as well as pediatric ganglioglioma and medulloblastoma, we found an association between ECMhi subtype and decreased patient survival (Fig. 1d), suggesting that ECM expression signature can be prognostic of clinical outcome in a subset of CNS tumors. In GBM, however, we detected an association in only one of the cohorts analyzed (Fig. 1d), indicating that ECM may not be prognostic of patient survival at baseline in GBM.
Next, we sought to determine if differences between ECMhi and ECMlo GBM are driven by changes in ECM gene expression alone, or by other gene expression programs that could induce ECM gene expression changes as a bystander effect. First, ECM subtypes were applied to primary and recurrent tumors from the longitudinal Glioma Longitudinal AnalySiS (GLASS) cohort. We did not observe any association between ECM subtype and tumor recurrence (Supplementary Fig. 1j), contrary to previous findings that ECM deposition is higher in recurrent GBM tissue23. Next, we performed differential expression analysis between primary ECMhi and ECMlo tumors. Gene ontology (GO) enrichment analysis of differentially expressed genes revealed an upregulation of immune effector process, wound healing, and angiogenesis pathways in adult glioma ECMhi tumors (Fig. 1e). In contrast, ECMlo samples were characterized by upregulation of neurosynaptic pathways (Fig. 1e), likely suggesting an enrichment of non-neoplastic cells. Additionally, ECMhi tumors upregulated numerous immune and stromal-related signatures (Fig. 1f).
Using data from the Ivy Glioblastoma Atlas Project (Ivy-GAP), we confirmed that ECMlo signature was preferentially expressed at the leading edge (LE) and infiltrative region (IT) of the tumor, which are known to consist largely of non-neoplastic cells24, whereas the ECMhi signature was upregulated in regions corresponding to microvascular proliferation (CTmvp) and hyperplastic blood vessels (CThbv), in line with a recent study25 (Supplementary Fig. 1l). We also examined in situ hybridization (ISH) and adjacent hematoxylin and eosin (H&E) tissue sections annotated for the same histologic features. We found that ECMhi hallmark genes COL1A1, COL4A1 were expressed in CTmvp regions (Supplementary Fig. 1m), suggesting that ECMhi signature is spatially associated with GBM vasculature.
ECMhi tumors are characterized by the presence of perivascular fibroblasts whose enrichment predicts poor response to immunotherapyThe vascular microenvironment is an important brain tumor niche with a heterogeneous and not fully revealed cellular makeup26. In order to identify vascular/perivascular cellular components contributing to the ECM stroma in CNS tumors, we applied SCIPAC – a tool designed to identify phenotype-associated cells in single-cell RNA-sequencing (scRNA-seq) data – to a scRNA–seq dataset from 16 GBM patients (Supplementary Fig. 2a–e)27. SCIPAC predicted 43% of PDGFRβ + ACTA2+ mural cells to be associated with the ECMhi signature (Fig. 2a). We sub-clustered and annotated mural cells based on gene expression signatures characterizing previously identified perivascular cell types in the human brain9. We identified two clusters as perivascular fibroblasts (P-FB; FBLN1, LAMA2) and meningeal fibroblasts (M-FB; SLC4A4, KCNMA1), as well as separate clusters of pericytes (PC; PDGFRB, COL4A1), and smooth muscle cells (SMC; ACTA2) (Fig. 2b). Integration of GBM mural cells with those from a human brain vascular atlas9 resulted in alignment of respective subpopulations in UMAP space (Fig. 2c). We found that 86% of perivascular fibroblasts were associated with the ECMhi phenotype, suggesting their pro-fibrotic role in the GBM TME (Fig. 2c). Additionally, we found that perivascular fibroblasts resembled previously identified murine brain fibroblast-like cells28 (Supplementary Fig. 2i). To determine the contribution of these perivascular cells to ECMhi and ECMlow states, we deconvoluted bulk gene expression profiles using a set of signature genes identifying each perivascular cell type. We found that ECMhi metagene was highly correlated to perivascular fibroblast, pericyte, and SMC signatures but not to meningeal fibroblast signature (Fig. 2g), likely reflecting their low frequency in GBM, which is consistent with rare cases of primary extracerebral meningeal GBM29. Notably, the P-FB cluster expressed several classical CAF markers such as PDGFRA, PDGFRB, COL1A1, and FAP17,30,31. Strikingly, we found that enrichment of perivascular fibroblasts was prognostic of poor clinical outcome in GBM (Supplementary Fig. 2g) and correlated with mesenchymal subtype (Supplementary Fig. 2f), suggesting their possible pro-tumorigenic role. Therefore, we refer to this population of cells as “CAF-like” whenever appropriate to reflect their CAF-like tumor-promoting phenotype.
Fig. 2: Cancer associated fibroblast-like cells of perivascular origin are present in ECMhi GBM and predict poor response to immunotherapy.
a Top: a UMAP plot showing subpopulations of mural cells. Bottom: barplot showing the frequency of ECMhi-associated cells within each cell type. b Marker genes identifying subpopulations of mural cells in GBM. P-FB – perivascular fibroblast; PC – pericyte; SMC – smooth muscle cell; M-FB – meningeal fibroblast. c Joint UMAP embedding of mural cells from a human brain vascular atlas (grey) and GBM mural cells (red). d UMAP embedding of GBM mural cells, colored by the enrichment of the perivascular fibroblast metagene (top) and SCIPAC predictions (bottom). e SCENIC-inferred regulon activity in subpopulations of mural cells. Shown in red are top 10 regulons for each mural cell subpopulation based on regulon specificity score (RSS). f RSS of transcription factors in perivascular and meningeal fibroblasts; 10 transcription factors with the highest RSS are labeled in red. g Pearson correlation between ECMhi/ECMlo metagenes and mural cell subpopulations. h Kaplan–Meier plot of overall and progression free survival for patients with high (red) or low (blue) presence of perivascular fibroblasts in Cloughesy (n = 28) and Zhao (n = 38) cohorts treated with anti-PD1 therapy (P values were calculated using log-rank test). i UMAP embeddings of scRNA-seq data from different tumors, colored by cell type (top) and positive (red) or negative (blue) association with ECMhi state, as predicted by SCIPAC. HGPT – high-grade pediatric tumors; LGPT – low-grade pediatric tumors.
Next, we applied SCENIC to infer active regulators of the fibroblast populations and confirm their identity in GBM. For perivascular and meningeal fibroblast populations, SCENIC predicted a high activity of several common brain fibroblast transcription factors ZIC1, FOXC1, NR2F232, TWIST133, and a CAF-specific transcription factor NR2F1 (Fig. 2f)34. The activity of these regulons was restricted to cells with perivascular and meningeal fibroblast signatures, indicating that their phenotype is distinct from that of pericytes or smooth muscle cells (Fig. 2e). These results suggest that fibrotic scarring in GBM may share some of its mechanisms with other CNS pathologies in which fibrogenic cells are derived from perivascular fibroblasts in response to inflammatory stimuli. Interestingly, however, our analysis of genomic copy number using the CopyKat algorithm identified a fraction of perivascular stromal cells as aneuploid (Supplementary Fig. 2d). Since mesenchymal differentiation of glioma stem cells (GSCs) into pericytes has been previously shown (deCarvalho et al. 2010; Ricci-Vitiani et al. 2010), these findings raise an intriguing possibility that malignant cells can undergo a mesenchymal differentiation to assume a CAF-like phenotype in GBM.
Next, to identify the presence of closely related stromal cell types in other ECMhi-enriched CNS tumors, we analyzed scRNA-seq data from neurofibroma (n = 3), meningioma (n = 7), low-grade pediatric tumors (LGPT; n = 26), high-grade pediatric tumors (HGPT; n = 23) and Bmets (n = 15) (Fig. 2i). Stromal/mesenchymal cells were detected at the highest frequency in BMets (21%), meningioma (26%), and neurofibroma (56%). We did not detect any stromal cells in HGPTs which our previous analysis identified to be ECMhi-enriched which could be ascribed to variability in cell isolation protocols, since detachment of cells embedded in the basement membrane requires stronger tissue dissociation methods28,35.
In order to verify their fibrogenic phenotype, we applied SCIPAC on scRNA-seq and tumor-matched bulk RNA-seq data. SCIPAC predicted fibroblasts as the cellular source of ECM in neurofibroma and meningioma (Fig. 2i). In Bmets, in addition to perivascular stromal cells, myeloid cells and endothelial cells were significantly associated with ECMhi phenotype, suggesting their possible role in ECM remodeling. No ECMhi-associated cells were found in HGPT and LGPT tumors, consistent with the absence of stromal cell types in these datasets. Next, in order to identify conserved stromal cell states across different cancers, we performed the mutual nearest neighbors (MNN) batch correction on combined data and sub-clustered stromal cells based on MNN-corrected gene expression values (Supplementary Fig. 2j, k). The majority of GBM perivascular fibroblasts were clustered together with mesenchymal stem-like cells (MSCs) from Bmets. Indeed, perivascular CAF-like cells also upregulated mesenchymal progenitor markers CTHRC1 and ISLR (Fig. 2d)36, indicating that MSCs may be a source of CAF-like cells in GBM, as previously suggested14. Neurofibroma fibroblasts, which have been characterized as distinct from classical fibroblasts37, formed a separate cluster. Clusters 3, 4, and 5 predominantly contained meningioma mesenchymal subtypes together with meningeal GBM fibroblasts; cluster 0 consisted almost entirely of pericytes and smooth muscle cells. Overall, the results of this integrative analysis suggest that despite their common role in ECM remodeling of the tumor stroma, stromal cells exhibit distinct and largely non-overlapping phenotypes across different CNS malignancies.
Finally, to determine whether perivascular fibroblasts play a role in anti-tumor immunity and response to immunotherapy in GBM, we interrogated retrospective data from two recent anti-PD1 immunotherapy trials21,22. In both cohorts, we found a significant reduction in overall survival for patients whose tumors were enriched in perivascular fibroblasts (Fig. 2h), but not other mural cell subpopulations (Supplementary Fig. 2h), suggesting that the presence of perivascular fibroblasts is prognostic of a poor immunotherapeutic response.
Glioblastoma perivascular fibroblasts express chemotactic factors that may recruit tumor-associated macrophages to the TMENext, we hypothesized that GBM perivascular fibroblasts may play a role in modulating neuroinflammation and contribute to the establishment of an immunosuppressive TME that is resistant to immune checkpoint blockade such as anti-PD1 antibodies. To verify this, we first performed cell type deconvolution of GBM bulk gene expression profiles. We found a strong positive correlation between the presence of perivascular fibroblasts and myeloid cells, including macrophages, dendritic cells, and monocytes (Fig. 3a). Perivascular fibroblast signature was positively correlated to expression of CD11B (ITGAM) and CD163, but not CX3CR1 (Fig. 3b), indicative of increased numbers of monocyte-derived macrophages38, as well as T cell exhaustion markers (Supplementary Fig. 3a). This was supported by differential abundance analysis of GBM scRNA-seq data, in which we found an enrichment of two populations of tumor-associated macrophages (TAMs) s-Mac1 and s-Mac2 (Supplementary Figure 2b), myeloid-derived suppressor cells (MDSCs) and proliferating (Prolif.) macrophages in samples that were classified as ECMhi based on pseudobulk expression profiles (Fig. 3c, Supplementary Fig. 2b, c). Additionally, macrophages, dendritic cells (DCs) and MDSCs displayed a lower expression of genes encoding MHC class II (MHC-II) molecules (Supplementary Fig. 3b), indicating their poor antigen-presenting capacity, characteristic of the M2-like macrophage state.
Fig. 3: Perivascular fibroblasts express chemotactic factors that may recruit tumor-associated macrophages to the GBM TME.
a Correlation between perivascular fibroblast (P-FB) signature score and monocyte-derived macrophage (ITGAM, CD163) and microglial (CX3CR1) marker genes. Value of the spearman correlation coefficient is shown together with the p value. b Spearman correlation between perivascular fibroblast signature score and immune cells fractions in bulk RNA-seq data, estimated using the TIMER algorithm. c UMAP embedding of myeloid cells colored by cell subpopulation. d Differential abundance analysis of ECMhi and ECMlo tumors. The left panel shows a graph of cellular neighborhoods superimposed on the UMAP embedding of the data. Color represents log fold change (logFC) relative to ECMlo, node size represents the size of the neighborhood, and edge width represents the amount of overlap between neighborhoods. Relative changes which were not statistically significant (p > 0.05) are not shown. The right panel shows the distribution of differential abundance of cellular neighborhoods across myeloid subpopulations. e Heatmaps showing interaction strength between P-FB cells and myeloid cells for select pathways. Mac – macrophage; Mic – microglia; Prolif. – proliferating macrophage; MDSC – myeloid-derived suppressor cell; DC – dendritic cell. f Expression of selected ligands and their receptors for pathways from (e).
To investigate possible mechanisms of macrophage recruitment by perivascular fibroblasts, we applied the CellChat algorithm that can infer cell-state specific signaling communications from scRNA-seq data39. CellChat predicted active chemoattractant signaling from perivascular fibroblasts to macrophages via chemokines CCL2, CXCL1, CXCL2, CXCL12, CSF1, and matricellular protein periostin (POSTN), which are known to induce chemotaxis and alternative polarization of tumor-supporting M2-like myeloid cells (Fig. 3d–f)3,40,41,42,43,44. These results suggest that perivascular fibroblasts may contribute to the establishment of an immunosuppressive microenvironment by driving M2-like TAM recruitment and polarization via chemotaxis and periostin signaling, respectively.
Perivascular fibroblasts promote immune-evasive stem-like cancer cell phenotype in GBMGSCs are maintained within perivascular collagen-rich niches45,46, and are known to evade antitumor immune responses through various mechanisms, including downregulation of MHC class I, induction of quiescence, and other mechanisms that promote immune tolerance47. In GBM, CAFs are known to enrich GSCs14. Therefore, we hypothesized that GBM perivascular fibroblasts may also modulate anti-tumor immune responses through maintenance of GSCs in the perivascular niche. In order to verify this, we first quantified the stem cell-like tumor phenotype by computing “stemness score”48. We found that ECMhi tumors displayed a higher stemness score across all three datasets analyzed (Fig. 4a), suggesting that a fibrotic microenvironment may favor the emergence and/or maintenance of glioma stem-like cell phenotype. We then classified glioma cells into oligodendrocyte-progenitor-like (OPC-like), neural-progenitor-like (NPC-like), astrocyte-like (AC-like), and mesenchymal-like (MES-like) cell states49, and found that ECMhi tumors were characterized by an enrichment of MES-like cells and reduced frequency of NPC-like and OPC-like states (Fig. 4b, Supplementary Fig. 3d), which is in line with the preponderance of myeloid cells in ECMhi tumors and their proposed role in maintaining the MES-like cell state49.
Fig. 4: Perivascular fibroblasts secrete factors that upregulate stem-like programs in ECMhi glioblastoma.
a Stemness signature scores in ECMhi and ECMlo tumors in TCGA (n = 175), CGGA325 (n = 325) and CGGA693 (n = 693) GBM cohorts. b Two-dimensional butterfly plot of glioma cell states in ECMhi and ECMlo tumors, colored by enrichment of the mesenchymal glioma stem cell (mGSC) signature. OPC – oligodendrocyte progenitor cell-like; AC astrocyte-like; NPC – neural progenitor cell-like; MES – mesenchymal-like. c Proneural (top) and mesenchymal (bottom) glioma stem cell scores in ECMhi and ECMlo tumors. Two-sided t-test. Benjamini-Hochberg adjusted P values are shown. d Strength of outgoing and incoming signaling in perivascular fibroblasts (P-FB) and MES-like glioma cells for selected pathways with known role in cancer stem cell maintenance. e Expression of ANGPTL4 and its receptors in GBM scRNA-seq data. In (a) and (c), p values were obtained using two-tailed t-test. P values corrected for multiple comparisons using the Holm–Bonferroni method are shown. For violin plots in (a) and (c), the center lines represent the median. The lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles, respectively). The upper whisker extends from the hinge to the largest value no further than 1.5 times of inter-quartile range (IQR) from the hinge. The lower whisker extends from the hinge to the smallest value at most 1.5 times of IQR from the hinge.
GBM contains hierarchies of mesenchymal and proneural GSCs (mGSCs and pGSCs, respectively) that are considered largely responsible for cancer cell heterogeneity observed within GBM tumors50. To understand the contribution of these progenitor states to glioma cell heterogeneity observed in ECMhi and ECMlow subtypes, we scored single cells using previously identified mGSC and pGSC signature genes. We found that MES-like glioma cells in ECMhi tumors were enriched in mGSCs (Fig. 4b, S4e), which is in line with their increased frequency.
Next, to elucidate possible mechanisms of mGSC enrichment, we compared CellChat-inferred signaling networks between ECMhi and ECMlo tumors. We found an upregulation of ANGPTL and PDGF signaling by perivascular fibroblasts to glioma cells, which have been implicated in maintenance of stem-like cell phenotype in GBM (Fig. 4d)51,52,53. MES-like glioma cells expressed multiple receptors for ANGPTL4, including SDC2 SDC3, SDC4, and integrins ITGA5 and ITGB1 (Fig. 4e), suggesting that the ANGPTL pathway may be responsible for the enrichment of GSCs in ECMhi tumors.
Comments (0)