Patients
Copy gain of BRCA1 exons 8–20 were identified in routine diagnostic setting in each family by Next-Generation Sequencing performed in four distinct medical centres. Exonic copy gains were confirmed by Multiplex Ligation-dependant Probe Amplification (MLPA, MRC Holland probe mix P002-D1) on DNA extracted from blood.
All patients provided informed consent and were included in the COVAR (COsegregation of VARiants) study (NCT01689584), authorized by Ethics Committee in 2011 (Comité de protection des personnes Ile de France III, Am5677-1-2940). All procedures involving human participants were conducted in accordance with the Declaration of Helsinki and its amendments.
DNA and RNA extraction
Blood DNA was extracted with the QiaSymphony DSP DNA Midikit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
RNA was extracted from B lymphoblastoid cell lines established by in vitro infection with Epstein Barr Virus. Cells were treated by puromycin to inhibit nonsense mediated decay. After storage in 1 mL Trizol (Invitrogen, ref. 15596026), RNA was extracted using the standard chloroform/isopropanol procedure.
Family analysis
Haplotype determination was performed by amplifying five microsatellite regions surrounding BRCA1 gene: D17S1327 downstream BRCA1 in the genome (5’-mCTAAGGAGGTTTCTCTGGAC-3’, 5’-TTCACAACTCAAGGTAAGATAGG-3’), D17S1323 in intron 12 (5’-mTAGGAGATGGATTATTGGTG-3’, 5’-AAGCAACTTTGCAATGAGTG-3’), D17S1322 in intron 19 (5’-mGCAGGAAGCAGGAATGGAAC-3’, 5’-CTAGCCTGGGCAACAGAACGA-3’), D17S855 in intron 20 (5’-mACACAGACTTGTCCTACTGCC-3’, 5’-GGATGGCCTTTTAGAAAGTGG-3’) and D17S1185 upstream (5’-mGGTGACAGAACAAGACTCCATC-3’, GGGCACTGCTATGGTTTAGA-3’). PCR was performed with AmpliTaq GOLD DNA Polymerase according to the manufacturer’s recommendations (Applied Biosystems, ref. 4311818) for 30 cycles (Hybridization: 55 °C). Amplified DNA (2μL) was then mixed with 0.5μL 500 LIZ dye size standard (Applied Biosystem, ref. 4322682) for fragment size determination by capillary electrophoresis.
As previously described4, likelihood ratios (LR) for pathogenicity were computed from clinical and histopathological family histories19,20, as well as on cosegregation analysis using a Bayesian statistical model described by Thompson et al. and updated by Belman et al.21,22. Probability for pathogenicity was computed using the multifactorial model defined by Goldgar et al.23. As prior probabilities of pathogenicity have not been calibrated for complex structural variants3,24, we tested prior probabilities ranging from 0.5 (prior odds = 1, so the posterior odds equal the LR, making the posterior probability depend only on the LR25) to 0.1 (a conservative prior).
Short-read and long-read DNA sequencing (DNA-seq)
Short-read DNA-seq was performed on a NextSeq 500 (Illumina) after enrichment using a custom SureSelect QXT kit (Agilent), as described previously8. Mapping on GRCh37/hg19 was performed with Bowtie2.
Long-read DNA-seq was performed for the first run on a Oxford Nanopore Technologies Minion Flow Cell R9.4.1 (ref. FLO-MIN106D) after library preparation by manufacturer ligation kit (Oxford Nanopore Technologies, ref. SQK-LSK110) on 2 μg DNA, as described previously8. For the second run, a long-read DNA library was prepared using the new chemistry with the long sequencing kit SQK-LSK114 on 2 µg DNA, as per the supplier’s recommendations. The DNA library was then injected into a Minion Flow Cell R10.4.1 (ref. FLO-MIN114). Computational enrichment was performed by adaptive sequencing (GRCh37/hg19). The first run targeted coding sequences of 120 genes including BRCA1 (49 Mb)8. The second run targeted the whole long arm of chromosome 17 (84 Mb) including BRCA1. Bioinformatics analysis was performed with a custom NanoCliD pipeline (https://github.com/InstituteCurieClinicalBioinformatics/NanoCliD) including Minimap2 for alignment on GRCh37/hg19, GRCh38/hg38, or T2T-CHM13/hs1.
Optical Genomic Mapping (OGM)
Ultra-high-molecular weight DNA was isolated and purified using the Bionano Prep SP-G2 Blood and Cell Kit as per the manufacturer’s instructions. Direct DNA labelling on CTTAAG sequence was conducted according to the DLS-G2 protocol with the DLE1 enzyme.
Labelled molecules were linearized into Saphyr chip G3.3 nanochannels to allow simultaneous direct imaging on the Saphyr instrument. A de novo assembly was carried out using the Bionano serve 3.7 and Access software version 1.7.
Genomic DNA and complementary DNA (cDNA) Sanger Sequencing
Breakpoints were confirmed by DNA Sanger sequencing after PCR amplification with primers 5’-GCTGTTTGCGTTGAAGAAGT-3’ and 5’-CTGCCATTTCTTTTCACTCTGG-3’ for breakpoint 1 (BP1); and 5’-ACCCCAGCACTCCTAAGAAC-3’ and 5’-GGGACCACTATCAGCTGACT-3’ for breakpoint 2 (BP2).
For RNA Sanger sequencing, RNA was reverse-transcribed using SuperScript II reverse-transcriptase (Invitrogen, ref. 18064014) as per the manufacturer’s instructions, with 1U/μL RNAse inhibitor (Applied Biosystems, ref. N8080119) and 2.5 μM Random Hexamer Primers (Invitrogen, ref. N8080127). cDNA was then amplified using a forward primer specific to BRCA1 exon 20 (5’-AGAAACCACCAAGGTCCAAAG-3’) and a reverse primer specific to BRCA1 exon 9 (5’-GCCTTATTAACGGTATCTTCAG-3’).
PCR reactions were performed with Taq DNA Polymerase (VWR, ref. 733–1301) as per the manufacturer’s instructions over 35 “touchdown” cycles (Hybridization: 58 °C x2; 57 °C x2; 56 °C x2; 55 °C x3; 54 °C x3; 53 °C x; 52 °C x4; 51 °C x5; 50 °C x10). Sequencing reactions were performed using Big Dye Terminator as per the manufacturer’s instructions (ThermoFisher, ref. 4337452).
Strand-specific short read RNA sequencing (RNA-seq)
Strand-specific RNA-seq was performed on a NextSeq 500 (Illumina) after library preparation with custom SureSelect XT HS2 RNA probes (Agilent). We followed the manufacturer’s protocol for strand-specific library preparation. Briefly, after initial preparation and fragmentation of 200 ng RNA, first-strand and second-strand cDNA were synthesized in two distinct steps with two distinct mixes. The second-strand cDNA mix contained dUTPs for specific second-strand marking. Reads were mapped on GRCh37/hg19 using STAR.
The sequencing depths of forward-strand and reverse-strand transcripts were compared to a merged bam file containing data from 12 distinct controls. These controls were patients suspected of carrying a genetic variant causing a splicing defect in a gene involved in paediatric cancer predisposition (n = 2), ataxia-telangiectasia or ataxia-telangiectasia-like disorders (n = 3), or digestive cancer predisposition (n = 7). All controls had provided informed consent for genetic analysis for diagnostic and research purposes.
Long-read direct RNA-seq
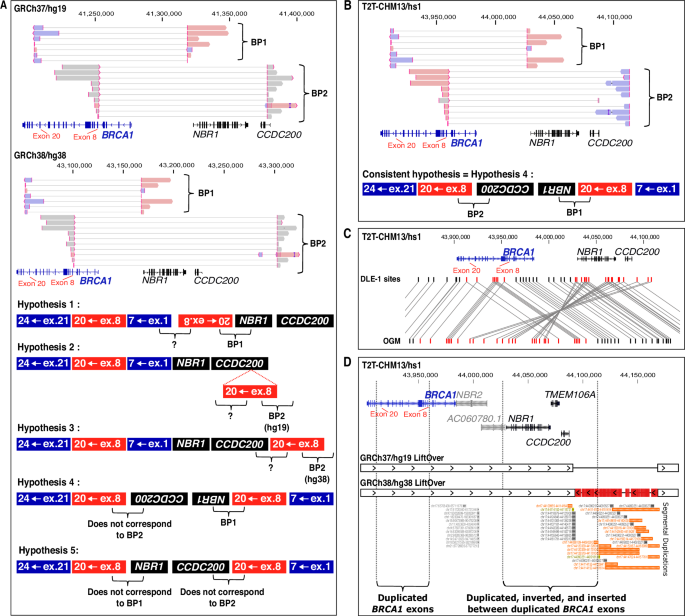
Long-read RNA-seq libraries were prepared from 1 µg of total RNA using the Oxford Nanopore Direct RNA Sequencing Kit (ref. SQK-RNA004). After an initial hybridization step, polyadenylated (polyA) messenger RNAs were captured, reverse-transcribed, and sequencing adaptors were ligated. Sequencing was performed using PromethION flowcell RNA (Oxford Nanopore Technologies, ref. FLO-PRO004RA) and reads were mapped to GRCh37/hg19 using Minimap2. For Fig. 2D design, alignments were displayed in Integrative Genomics Viewers (IGV 2.15.4) software, and fusion reads spanning BRCA1 were exported for manual reconstruction on BP1 structure.
Reference transcripts
Represented transcripts correspond to NM_007294.4/ENST00000357654.9 for BRCA1, ENST00000657841.1 for NBR2, NM_005899.5/ENST00000590996.6 for NBR1, NM_145041.4/ENST00000612339.4 for TMEM160A, NM_001363254.2/ENST00000636331.2 for CCDC200, and ENST00000635600.1 for NBR1 antisense transcript AC060780.1 (sharing the same first exon as NR_110868/ LOC101929767).


Comments (0)