Pharmacology analysis, synthetic accessibility, and novelty of the developed compound prediction
Following the design of compounds, 1A and 1B, SciFinder was employed to predict the novelty of the two compounds. The pharmacological and pharmacokinetic properties were evaluated according to Lipinski's rule of 5, encompassing an ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) test conducted through in-silico analysis using the SwissADME and pkCSM online tools. In addition, the SwissADME web interface was used to assess the synthetic accessibility of the compounds [13,14,15].
ChemistryGeneral information
The starting materials and catalysts used in the chemical synthesis, as well as the chemicals for buffer preparations, antibodies, and fluorescent dyes in this study, were purchased from various suppliers. All commercially available reagents were used as received. Reaction progress was tracked by analytical thin layer chromatography (TLC) on silica gel plates, visualized under ultraviolet (UV) light at 254 and 366 nm. Compound purities were evaluated using high-performance liquid chromatography (HPLC) with a Waters Alliance system and Shimadzu equipment using a suitable mobile phase on a Waters C18 column, and through melting point determination with a digital capillary apparatus. High-resolution mass spectra (HRMS) were obtained with an Agilent 6520 Q-TOF. Fourier-transform infrared (FTIR) spectra were recorded with a Shimadzu FTIR-8310, and 1H and 13C nuclear magnetic resonance (NMR) spectra were acquired using a Bruker Ascend instrument.
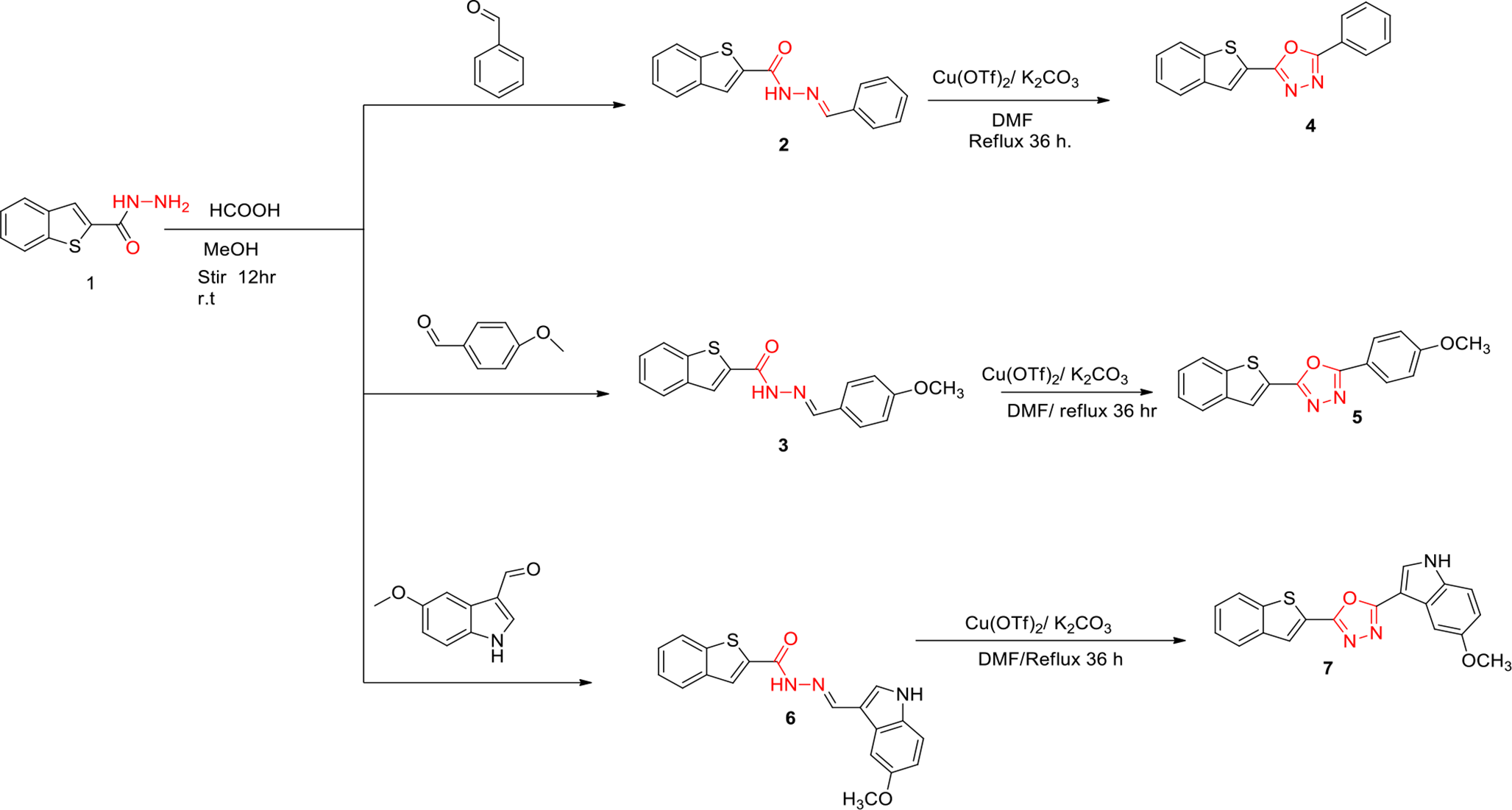
Synthesis and characterization of N-hydroxy-2,2′-bipyridine-6-carboxamide (1A)
5.5 mL of 30% hydrogen peroxide was added to 5 g (32 mmol) of 2,2′-bipyridine in 25 mL of trifluoroacetic acid. The reaction was stirred for 2 h followed by neutralization using 6 N sodium hydroxide and extracted with chloroform (3 × 150 ml) to obtain 2,2′-bipyridine-N-oxide. Subsequently, the reaction mixture of 3 g (17 mmol) of 2,2′-bipyridine-N-oxide in 30 mL of dichloromethane under nitrogen atmosphere was cooled to 0 °C and 8.72 mL (69 mmol) of trimethylsilyl cyanide and 4.08 mL of benzoyl chloride (34 mmol) were added to the reaction mixture, which was then maintained at room temperature with continuous stirring for 16 h to yield 2,2′-bipyridine-6-carbonitrile. 500 mg (2.7 mmol) of 2,2′-bipyridine-6-carbonitrile was then added with 10 mL of ethanol and 25 mL of sulfuric acid and the mixture was heated to 70–80 °C and stirred for 36 h to yield ethyl 2,2′-bipyridine-6-carboxylate. Finally, 0.4 g (1.7 mmol) of ethyl 2,2′-bipyridine-6-carboxylate, 0.182 g (2.6 mmol) of hydroxylamine hydrochloride, and 0.628 mL (3.5 mmol) of N,N′-diisopropylethylamine were combined at room temperature. The reaction mixture was heated to 75 °C and stirred for 48 h and the resulting crude product was purified by column chromatography and the product was eluted with 5–7% of methanol/dichloromethane.
White solid; mp: 107–112 °C; Purity 99.59% (HPLC); Yield: 33%; FTIR (KBr, cm−1): 3223.05 (NH, OH); 1664.57 (CO); 1573.91 (NH); 1257.59 (CN). 1H NMR (400 MHz, DMSO-d6, ppm): δ 11.70 (s, 1H, hydroxamic NH); 9.26 (s, 1H, hydroxamic OH); 8.94 (d, 1H, J = 7.6 Hz); 8.71 (d, 1H, J = 4.4 Hz); 8.56 (d, 1H, J = 7.6 Hz); 8.12 (t, 1H, J = 8.0 Hz); 8.04 (d, 1H, J = 7.2 Hz); 7.98 (t, 1H, J = 8.0 Hz); 7.49 (t, 1H, J = 5.8 Hz). 13C NMR (100 MHz, DMSO-d6, ppm): δ161.53; 154.86; 154.80; 150.03; 149.61; 139.25; 137.73; 125.09; 123.08; 122.39; 122.24. HRMS (Q-TOF) m/z calculated for C11H9N3O2, [M+H]+ 216.0768 g/mol. Found, 216.0334 g/mol. The spectral data of intermediates and compound 1A is given in the supplementary file.
Synthesis and characterization of 1-([2,2′-bipyridin]-6-yl)-3-hydroxyurea (1B)
4 g (23 mmol) of 2,2′-bipyridine-N-oxide was dissolved in 40 mL of 2,2,2-trifluoro toluene. The reaction mixture was then cooled to 0 °C, followed by the sequential addition of 12.2 mL (11.6 mmol) of tertiary butylamine and 13.64 g (69 mmol) of p-toluenesulfonyl chloride. After stirring for 18 h, 200 mL of trifluoroacetic acid was added, and the mixture was heated to 90 °C for 24 h. Trifluoroacetic acid was then removed, and the resulting compound was acidified with 6 N hydrochloric acid (pH 2–3) and then washed with methyl tertiary butyl ether: ethyl acetate (50 mL: 50 mL). The aqueous layers were then basified with 6 N sodium hydroxide (pH 10–12) to get (2,2′-bipyridin)-6-amine.
In a separate reaction, 1.21 g (7.6 mmol) of O-benzyl hydroxylamine hydrochloride was added to a solution of 1.23 g (7.6 mmol) of 1,1′-carbonyldiimidazole and 1.3 g (7.6 mmol) of N,N′-diisopropylethylamine in dry tetrahydrofuran at room temperature under nitrogen (N2) atmosphere. After stirring for 30 min, this mixture was added to a solution of 650 mg (3.801 mmol) of (2,2′-bipyridin)-6-amine in 10 mL of tetrahydrofuran at room temperature under N2 atmosphere to 1-([2,2′-bipyridin]-6-yl)-3-(benzyloxy)urea. This compound was then subjected to hydrogenation with ethanol and palladium on carbon under a hydrogen pressure (H2) of 600 psi for 5 h. After completion of the reaction, the mixture was filtered over a celite bed and washed with 50 mL of ethanol. The crude product was then purified by column chromatography and eluted in 30–40% ethyl acetate-hexane.
White solid; mp: 167–173 °C; Purity 95.93% (HPLC); Yield: 45%; FTIR (KBr, cm−1): 3209.19 (NH, OH); 1657.89 (CO); 1555.75 (NH) 1244.53 (CN). 1H NMR (400 MHz, DMSO-d6, ppm): δ 9.46 (s, 1H, hydroxamic OH); 9.26 (s, 1H, amide NH); 8.76 (s, 1H, hydroxamic NH); 8.68 (d, 1H, J = 4.4 Hz); 8.31 (d, 1H, J = 8.0 Hz); 8.02 (d, 1H, J = 8.0 Hz); 7.93 (m, 3H) and 7.45 (t, 1H, J = 6.2 Hz), 13C NMR (100 MHz, DMSO-d6, ppm): δ157.66; 155.20; 154.02; 152.03; 149.76; 139.79; 137.75; 124.67; 120.90; 115.35; 112.82. HRMS (Q-TOF) m/z calculated for C11H10N4O2, [M + H]+ 231.0877 g/mol. Found, 231.0396 g/mol. The spectral data of intermediates and compound 1B is given in the supplementary file.
Biological evaluationCell culture
The human cancer cells, namely, SiHa (cervical), MCF7 (breast) and Cal27 (oral) were procured from the ATCC (Manassas, VA, USA). The normal human foreskin fibroblasts were isolated after obtaining the ethical clearance from the institutional ethics committee, Kasturba Medical College, MAHE, Manipal, India. Isolation of fibroblasts was performed using skin epidermis (human dermal fibroblasts (HDF). The cells were cultured in DMEM (Dulbecco's Modified Eagle Medium) with 10% FBS (Fetal Bovine Serum) (Himedia, India). The culture medium was then renewed every 48–72 h. The cells were cultured under optimum conditions by maintaining the cells in incubator (Eppendorf CellXpert® C170, Hamburg, Germany) with 5% carbon dioxide (CO2) and humidified air at 37 °C.
In-vitro cell viability assay
The cells were trypsinized using 0.25% trypsin, and their total count was determined using a haemocytometer. Subsequently, 10,000 cells/well for SiHa, Cal27, and MCF7 cells and 5000 cells/well for fibroblast cells were seeded in 96 well plate and incubated for 24 h in CO2 incubator at 37 °C. Then 5 mg/mL of 3[-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) was added after a 48 h incubation with varying concentrations of the test compounds. Post MTT treatment, the plates were incubated for an additional 4 h at 37 °C. Subsequently, dimethyl sulfoxide (DMSO) was introduced to each well to dissolve the formazan crystals. The optical densities (O.D) of the samples were measured at 570 nm and 640 nm using Tecan Spark (Tecan, Männedorf, Switzerland). The results were represented as a percentage of the control and were computed as inhibitory concentration (IC50) values utilizing GraphPad Prism 9 software. Each compound was put in triplicate, and each experiment was replicated thrice [16].
In-vitro wound healing assay
2 × 105 cells/well of Cal27 cells were seeded in 6 well plate to establish a monolayer. An artificial scratch was generated in the monolayer with a sterile 200 μL pipette tip which was followed by phosphate buffered saline (PBS) wash. Fresh culture medium, both without (control) and with the addition of test compounds, was then applied and incubated for 48 h. The closure of the scratch was observed and documented at 0, 24, and 48 h intervals using an inverted microscope (Zeiss, Neu-Isenburg, Germany). Scratch image analysis was conducted using ImageJ software (Wayne Rasband, USA). The progression of the open wound area percentage was graphically represented over time for each respective group.
Cell cycle analysis
1 × 106 cells/mL of Cal27 cells were seeded in 10 cm plate and incubated for 24 h. They were then synchronized through serum starvation for 24 h, followed by treatment with compounds at their respective IC50 values for 48 h in culture medium. Afterward, cells were collected via trypsinization (0.25% trypsin), fixed overnight in 70% ethanol at 4 °C, treated with RNase A (10 µg/mL), and stained with propidium iodide (50 μg/mL) for 30 min at room temperature in the dark. Cell cycle distribution was analyzed using a Partec CyFlow Space with FloMax software (Partec, United States of America) [17].
Apoptosis assay
1 × 106 cells/mL of Cal27 cells were seeded in 10 cm culture plates and allowed to grow till 70–75% confluency. They were then treated with compounds at their respective IC50 concentrations for 24 h. After treatment, the cells were washed twice with ice-cold PBS and incubated in the dark at room temperature in 100 mL of 1X binding buffer containing 1 µL Annexin V-fluorescein isothiocyanate (FITC) and 12.5 µL propidium iodide (PI). Following 15 min incubation, the percentage of apoptotic cells was analyzed using a Partec CyFlow Space with FloMax software (Partec, United States of America) [18].
Intracellular reactive oxygen species (ROS) measurement
1 × 106 cells/mL of Cal27 were seeded in 10 cm culture plates and allowed to grow for 24 h until they reached 70–75% confluency. They were then treated with compounds at their respective IC50 concentrations for 48 h. Followed by treatment with 1 µg/mL diacetyldichlorofluorescein (H2DCFDA) dye for 45 min at 37 °C. Fluorescence of oxidized dichlorofluorescein (DCF) was measured using a Partec CyFlow Space with FloMax software (Partec, United States of America) [19].
Anchorage dependent colony formation assay
Cells were seeded at a density of 500 cells per well in 6-well culture plates. After 24 h of incubation, cells were treated with the compounds at their respective IC50 concentrations and incubated for 2 weeks with a continuously changing medium every three days. On the 14th day, the culture media was removed, colonies were stained with 0.4% crystal violet for 10 min, then followed by a PBS wash twice until the excess stain was removed, and the stained colonies were counted.
HDAC activity assay
HDAC fluorometric activity assay was employed to measure in vitro HDAC inhibition. Enzymes (HDAC), test compounds (inhibitors), and substrates were all diluted using HDAC assay buffer. In brief, HDAC assay buffer was added into each well, followed by the addition of test compounds (10 μL) to the assay buffer at various concentrations. Subsequently, a nuclear extract containing HDACs was added, and the mixture was incubated for 30 min. The enzymatic reaction was initiated by introducing fluorogenic substrates Boc-Lys(Ac)-AMC (MAL). After 2 h of incubation under shaking conditions at 37 °C, the reactions were halted by adding a stop solution (10 mg/mL trypsin and 0.2 mM Trichostatin A (TSA) in assay buffer). Following an additional 15 min of incubation at 37 °C, fluorescence was measured at emission wavelength of 354 nm and excitation wavelength 450 nm using a Tecan Spark (Tecan, Männedorf, Switzerland). The fluorescence in wells without test compounds was considered as control with 100% enzymatic activity [20].
Molecular docking
The free binding energy (kcal/mol) for compound 1A and the standard suberoylanilide hydroxamic acid (SAHA) to various HDAC isoforms was determined, using Maestro, Schrödinger (Version—11.2) as mentioned by Pai et al., 2022 [21].
Molecular dynamic simulation
Molecular Dynamic (MD) simulation was conducted for compound 1A and the standard SAHA in complex with HDAC 2 protein to enhance our insights into the stability of their interactions. The Desmond module of Schrodinger was employed for the MD simulations, following a three-step workflow as mentioned by Pai et al., in 2022 [21].


Comments (0)