
Remember me

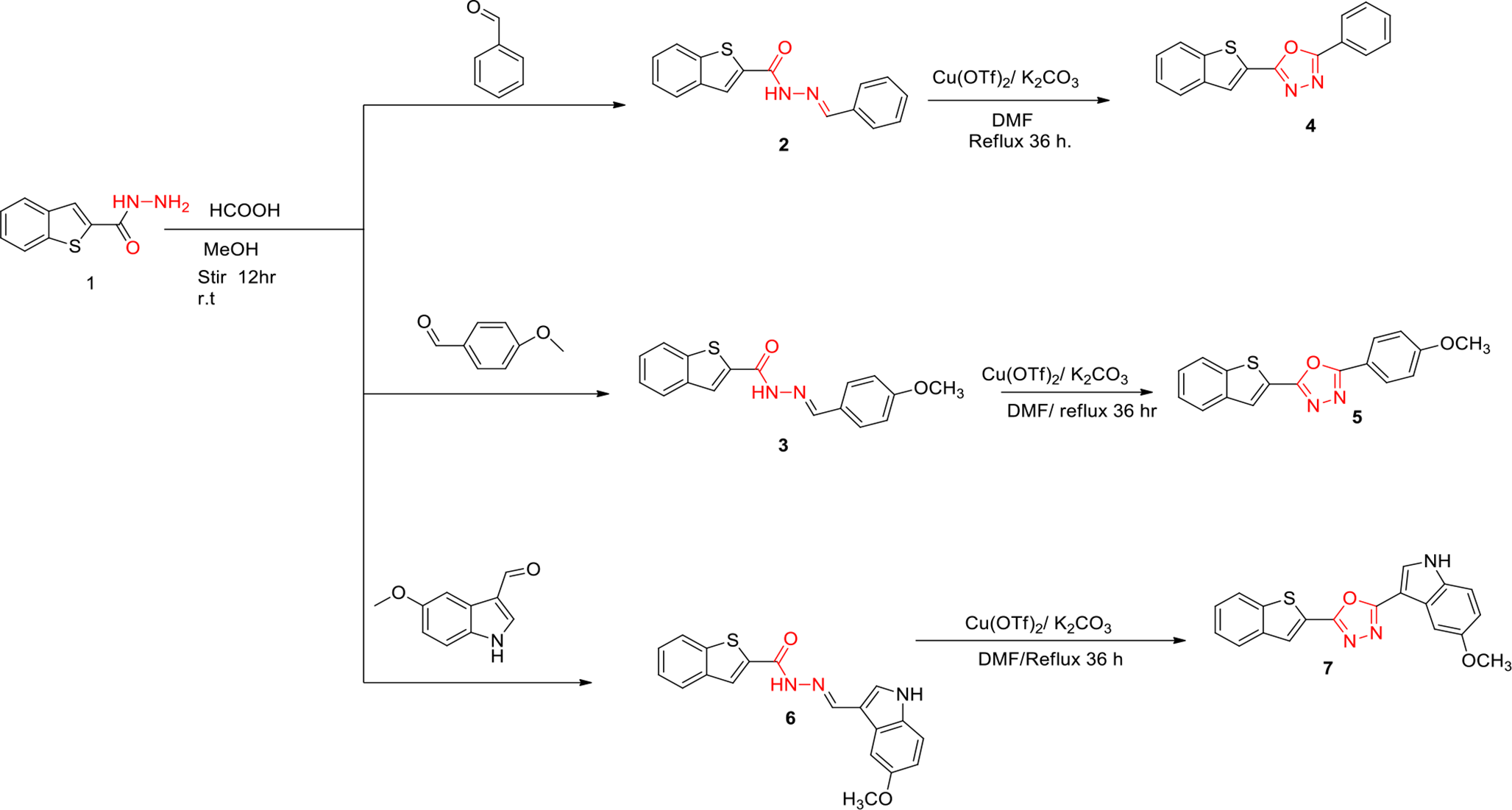
The vacuum-optimized geometry of vildagliptin (Fig. 1) closely aligns with its X-ray diffraction (XRD)-derived crystal structure. Selected geometric parameters are summarized in Table 1, with complete data provided in Table S1. The RMSD for interatomic distances between experimental and calculated values is 0.09 Å, indicating a strong correlation. As shown in Figure S1, the deviations primarily involve distances between hydrogen atoms and heavier atoms, which can be attributed to the nature of the XRD method, where the positions of lighter atoms are associated with greater uncertainty. The RMSD for bond angles between individual atoms is 1.11 degrees, indicating good agreement between the calculated and experimental data (Figure S2). The strong agreement between computationally derived and experimental structures underscores the robustness of the theoretical model, enabling detailed analysis of the compound’s geometric features. Minor discrepancies between DFT-optimized and XRD-determined geometries primarily involve bond lengths (e.g., O2-H23) and bond angles within non-cyclic amino, methylene, and carbonyl groups. These variations likely arise from structural relaxation in vacuum conditions and the absence of intermolecular interactions present in the crystalline environment.
Fig. 1
Optimized structure of vildagliptin and atom numbering
The carbon-carbon bond lengths within the adamantane scaffold align with literature-reported values for such bonds, averaging approximately 1.54 Å. Similarly, carbon-hydrogen bond lengths (≈ 1.09 Å) closely match theoretical predictions, though they exhibit a ~ 10% elongation compared to experimental data - a well-documented discrepancy in XRD studies.
The C16–C20 bond in the pyrrolidine ring is elongated relative to other C–C bonds in the system, likely due to steric and electronic effects arising from the substitution of a hydrogen atom at C16 with a nitrile moiety. Conversely, the C16–C21 bond length is shorter than typical single C–C bonds, attributed to electron density redistribution influenced by the adjacent nitrogen atom connected to C21.
Oxygen-carbon bond lengths further highlight structural specificity: the C9–O2 bond measures 1.433 Å, consistent with a single bond, while the C5-O1 bond (1.222 Å) reflects its double-bond character within the carbonyl group. The O2–H23 bond length (0.965 Å) aligns with standard values for hydroxyl-group interactions, corroborating the integrity of the hydrogen-bonding network in the optimized structure.
Most of the bonds connecting carbon and nitrogen atoms in the VILD molecule measure around 1.46 Å, with two notable exceptions: the C21–N6 and C5–N3 bonds. The former is a triple bond with an interatomic distance of 1.15 Å, while the latter is a single bond measuring 1.362 Å. The unusually short length of the C5–N3 bond can be attributed to an electron density shift from the carbon atom, induced by its adjacent double bond to a highly electronegative oxygen atom, thereby imparting a partial double-bond character.
Within the adamantane scaffold, most bond angles between carbon atoms are close to 109°, consistent with sp2 hybridization. An outlier is the angle defined by atoms C15-C18-C9, which measures 110.8°. The terminal atoms in this angle (C15 and C9) are bonded to heteroatoms - nitrogen and oxygen, respectively, whose lone pairs slightly perturb the ideal tetrahedral geometry of the neighboring carbon atoms. Furthermore, the C11–C9–O2 and C7–C9–O2 angles are approximately 111°, whereas the C18–C9–O2 angle is reduced to 106°.
The C13–N4–C15 bond angle (117.8°) deviates markedly from the idealized tetrahedral geometry, suggesting localized electronic perturbations. In contrast, angles within the amide functional group - C13–C5–O1 (≈ 122°), N3–C5–O1 (≈ 122°), and C13–C5–N3 (116°) - align with literature-reported values for amide-bonded systems, confirming the structural integrity of this motif.
Within the pyrrolidine ring, bond angles generally conform to literature values, with the exception of the C10–N3–C16 angle (112°), which exhibits slight expansion likely attributable to steric strain from adjacent substituents. Atoms C16, C21, and N6 are almost collinear, forming an angle of 178.2°, while the carbonyl and amino groups display near-coplanar alignment (N4–C13–C5–O1 dihedral angle: 11°).
Non-planarity of the pyrrolidine ring is evident: atoms C16–N3–C10–C19 lie nearly coplanar (dihedral angle: 6.3°), whereas C20 resides out-of-plane. Dihedral angles for C10–N3–C16–C20 (17.3°) and N3–C10–C19–C20 (27.4°) further underscore the ring’s puckered conformation. The nitrile group adopts a near-perpendicular orientation relative to the carbonyl moiety (C5–N3–C16–C21 dihedral angle: 87.2°), reflecting steric and electronic constraints imposed by the adamantane scaffold.
Table 1 Selected geometrical parameters of vildagliptin optimized in vacuumVibrational frequency analysisVildagliptin’s vibrational wavenumbers were analyzed by comparing experimental FTIR and FT-Raman spectra with DFT-simulated spectra. Theoretical spectra were scaled by a factor of 0.965 to improve alignment with experimental data, a standard correction to account for phase discrepancies (gas-phase calculations vs. solid-state measurements) [56]. Figure S3 presents the RMSD dependence of experimentally and computationally obtained peak wavenumbers on the scaling factor, varied from 1.000 to 0.900. The absence of imaginary frequencies in the computational output confirms the optimized geometry resides at a true potential energy minimum.
The FTIR spectra are partially discussed in the study [57], however, that manuscript provides only a superficial interpretation of the VILD FTIR spectrum and contains some rather obvious errors. The spectra are also addressed in the paper [58], but this work also omits most of the vibrational modes, such as those originating from the cyano group.
The molecule, exhibiting C1 symmetry, comprises 47 atoms and 135 fundamental vibrational modes. Each mode was optimized using algorithms in the VEDA 4 software, and its potential energy distribution (PED) was determined accordingly. Experimental and simulated FTIR spectra are presented in Fig. 2, while normalized Raman spectra are shown in Fig. 3. The spectra were not subjected to baseline correction. A comprehensive vibrational assignment, correlating observed wavenumbers with theoretical modes, is detailed in Table 2.
Fig. 2
Experimental A and theoretical, B FTIR spectra of vildagliptin
Fig. 3
Experimental A and theoretical, B Raman spectra of vildagliptin
The O–H stretching vibration of the hydroxyl group appears as a broad band in the FTIR spectrum, centered at 3385 cm−1. In the Raman spectrum, this mode manifests as a faint, low-intensity signal near the noise threshold. The broadening of the IR band likely arises from intermolecular hydrogen bonding within the crystalline lattice. The DFT-calculated wavenumber for this mode (3673 cm−1) exhibits a significant redshift compared to experimental data, a discrepancy attributable to the harmonic approximation used in simulations and the exclusion of crystal packing effects. In the literature, one can find interpretations of FTIR spectra for the core structure of vildagliptin (3-amino-1-adamantanol), where a characteristic broad band from the hydroxyl group can be observed around 3350 cm−1 [59]. This band partially masks the N–H stretching vibration, which appears as a sharp peak around 3300 cm−1 [60].
The COH bending vibration is observed as a broad IR band overlapping with HCCC torsional modes. In the Raman spectrum, this feature appears as a weak signal at 1354 cm−1. The OHC bending vibration also contributes to a combination band at 1102 cm−1 (PED: 16%). At 1055 cm−1, both FTIR and Raman spectra exhibit a broad band that lacks a corresponding feature in the computational model; this band likely arises from a combination vibration incorporating a COH bending component and is notably red-shifted due to intermolecular forces in the solid phase.
HOCC torsional vibrations lie beyond the FTIR detection range but were identified via Raman spectroscopy and computational analysis at 272 cm−1 (calculated: 273 cm−1) and 236 cm−1 (calculated: 246 cm−1).
The O–C stretching vibration of the hydroxyl group is observed as intense bands at 1032 cm−1 in both IR and Raman spectra. This mode also contributes to combination bands at 882 cm−1 (PED 10%) and 515 cm−1 (PED 11%). Bending and torsional modes involving OCC and OCCC moieties appear as components of multiple combination bands below 460 cm−1.
The prominent IR and Raman band at 1659 cm−1 is unambiguously assigned to the carbonyl (C = O) stretching vibration. OCC bending modes contribute to combination bands observed at 699 cm−1 (PED 15%) and 346 cm−1 (PED 11%). Torsional OCNC vibrations manifest as composite bands, likely corresponding to features at 984 cm−1 (PED 10%), 603 cm−1 (PED 21%), 586 cm−1 (PED 11%), and 551 cm−1 (PED 22%).
A medium-intensity, sharp IR band at 3293 cm−1, accompanied by a moderately intense Raman feature, is assigned to the N–H stretching vibration of the secondary aliphatic amine group, consistent with the characteristic spectral profile of such moieties. The HNC bending vibration of the amine group is confidently identified as the medium-intensity IR/Raman band at 1453 cm−1, while the HNCC out-of-plane bending modes appears as a distinct bands at 737 and 699 cm−1.
The N–C stretching vibration of the amine group manifests as a broad band at 1151 cm−1 and contributes to a combination mode at 1102 cm−1 (PED 13%). CNC bending vibrations are observed as low-frequency combination modes at 699, 480, and 432 cm−1. Torsional NCCC vibrations are tentatively attributed to features near 435, 24, and 13 cm−1.
The sharp, high-intensity IR and Raman band at 2239 cm−1 is assigned to the C≡N stretching vibration of the nitrile group - a feature seldom reported in organic compounds. Bending modes of this group contribute to low-frequency combination bands, tentatively identified at 495 cm−1 (calculated: 521 cm−1, PED 10%), 281 cm−1 (PED 24%), and 129 cm−1 (PED 16%). Torsional NCCH vibrations, lying beyond the instrumental detection range, are hypothesized near 224 cm−1 (PED 18%).
The N-C stretching vibration of the amide group appears as an intense IR band and weak Raman feature at 1406 cm−1. HCCN torsional modes correlate with a medium-intensity IR band at 984 cm−1, while low-intensity CCNC torsional vibrations manifest as components of combination bands below 500 cm−1. Bands at 1248 and 1174 cm−1 are partially attributed to N-C stretching involving pyrrolidine ring carbons. A weak IR band at 950 cm−1 is tentatively linked to the CNC bending vibration of the amide nitrogen atom within the pyrrolidine scaffold.
The C–H stretching vibrations generate overlapping medium-intensity IR bands and stronger Raman features within the 3012–2849 cm−1 range. Notably, the bands at 3012 and 2994 cm−1 are distinct, corresponding to CH stretching within the pyrrolidine ring, while those at 2957, 2952, 2932, and 2882 cm−1 are attributed to adamantane-associated C-H bonds. A band at 2848 cm−1 likely corresponds to methylene group C-H stretching.
Bending vibrations of HCH initiate near 1478 cm−1, with Raman spectra displaying a composite band spanning 1478–1373 cm−1, encompassing symmetric HCH bending modes of the methylene group, pyrrolidine ring, and adamantane scaffold. Overlapping bands at 1263–1280 cm−1 reflect HCH wagging vibrations, predominantly manifesting as combination modes. Raman bands at 1189, 1173, and 1054 cm−1 are linked to combination modes involving HCH bending contributions. In the ranges of 1269–998 cm−1 and 984–882 cm−1, bending vibrations of the twisting and rocking types, respectively, are observed as components of combination vibrations with a relatively low percentage of Potential Energy Distribution (PED). This is consistent with literature data obtained for similar compounds [61].
Computational modeling indicates torsional HCCC vibrations contribute to combination bands across a broad spectral range (1354–281 cm−1). However, their low potential energy distribution (PED) values and weak intensities suggest minimal energetic contributions to the overall vibrational profile.
The C-C stretching vibrations are observed in both the IR and Raman spectra with low intensity, beginning at 1263 cm−1 according to the computational model. However, in the experimental spectra these vibrations are assigned to a wavenumber of 1006 cm−1, and at lower wavenumbers they contribute to combination bands. CCC bending vibrations appear as components of combination bands, for example at 683, 635, and 585 cm−1. Additionally, CCCC twisting vibrations, which exhibit low intensity, have been detected experimentally only at 644, 461, and 346 cm−1. The remaining wavenumbers attributed to these modes through computational analysis are listed in Table 2.
Table 2 Experimental and calculated vibrational frequencies (cm− 1) with respective assignments and PED valuesHirshfeld surface and 2D fingerprint plotTo analyze the intermolecular interactions among vildagliptin atoms within the crystal lattice, a Hirshfeld surface analysis and 2D fingerprint plots were performed. Figure 4 displays the Hirshfeld surface using dnorm (normalized contact distance [62]), where the minimum contact distance (red) and the maximum contact distance (blue) are − 0.3669 and 1.6169, respectively. It is evident that the intermolecular interactions predominantly occur between nitrogen and oxygen atoms, as well as the hydrogen atom of the hydroxyl group.
Fig. 4
Hirshfeld surface of VILD mapped over dnorm
The 2D fingerprint histogram (Fig. 5) provides a more detailed breakdown of various intra- and intermolecular interactions along with their estimated percentage contributions. For example, interactions between the carbon atom of the nitro group and hydrogen atoms of neighboring molecules account for 4.9% of the total interactions. Interactions involving nitrogen atoms from the nitro and amino groups with hydrogen atoms comprise 17.8%, while those between oxygen atoms and hydrogen atoms account for 13.8%. The majority of the intermolecular interactions (60.8%) result from mutual interactions between hydrogen atoms. The remaining calculated parameters, such as distance to external atom, distance to internal atom, shape index, curvedness, and fragment patch, are presented in Table 3.
Fig. 5
2D finger plot graphs representing the number of molecular interactions of VILD. A– O H interactions, B– H–H interactions, C– N–H interactions, D– C–H interactions
Table 3 Hirshfeld surface properties of VILDMolecular electrostatic potential (MEP) surface analysisThe ESP surface analysis provides a visual representation and interpretation of the charge distribution around a molecule. This technique is instrumental in predicting how the molecule may interact with ions, reactive species, or solvents by highlighting regions predisposed to electrophilic or nucleophilic attacks. It also offers insights into the binding affinity of drug molecules toward proteins and their preference for polar or nonpolar solvents. The MEP map is constructed on a molecular surface defined by a specific electron density value [63].
For this study, the electrostatic potential was calculated using a molecular model optimized at the B3LYP-D3BJ/6-311++G(2d, p) level. The map indicates the most negative potential in red (−5.456 × 10− 2, −1.485 eV) and the most positive in blue (4.556 × 10− 2, 1.24 eV), with the color scale progressing from red to orange, yellow, green, and blue (Fig. 6). The N6 atom of the nitrile group, due to its lone electron pair, exhibits the most negative potential. Likewise, the oxygen atoms O1 and O2, each carrying two lone pairs, display high electron density and appear in red. These regions are key contributors to the electrophilic reactivity of vildagliptin with less electronegative species.
Fig. 6
Molecular electrostatic potential surface (MEP) of vildagliptin
Conversely, the highest positive potential is located on the outer part of the pyrrolidine ring, particularly around the C10-C19-C20 region. This area, depicted in light blue on the MEP map, is susceptible to nucleophilic attack due to the electron density being drawn toward nearby electronegative oxygen and nitrogen atoms. The adamantane ring shows a uniformly high potential, represented in green, except for the region with the attached hydroxyl group O2-H23, which creates localized areas of strong negative potential near the oxygen and strong positive potential near the hydrogen.
Nonlinear optical (NLO) propertiesThe exploration of non-linear optical (NLO) characteristics is a critical element in contemporary photonics and optoelectronics, driving the development of materials with specific optical functionalities. These properties arise from the non-linear response of a material’s electron density to high-intensity electromagnetic fields, diverging from the linear response described by classical optics. Density functional theory (DFT) offers a robust framework for predicting and analyzing NLO phenomena, yielding insights into the structure-property relationships essential for material design [64].
To assess the NLO potential of vildagliptin, dipole moment (µ), polarizability (α), and first hyperpolarizability (β) were computed using its vacuum-optimized geometry. The computed polarizability and hyperpolarizability of VILD were determined to be 31.8 × 10− 24 esu and 1.16 × 10− 30 esu, respectively, representing a considerable enhancement over urea - the prototypical NLO material (Table 4). These results suggest that the compound under investigation may be a promising candidate for further research into optically active materials.
Table 4 Calculated values of dipole moment, polarizability and hyperpolarizability for VILDNatural bond orbital (NBO) analysisNBO analysis offers valuable insights into the electronic structure and bonding features of molecules, detailing charge distribution, hybridization states, and donor-acceptor interactions that influence molecular stability and reactivity. By quantifying second-order perturbation energies (E(2)), this method identifies key hyperconjugative interactions and lone pair contributions, which are essential for understanding intramolecular charge transfer and non-covalent forces involved in ligand-receptor binding [65].
NBO calculations were performed on the gas-phase optimized structure using the same functional and basis set with dispersion correction. Data concerning the types of donor and acceptor orbitals, along with second-order perturbation energies exceeding 4 kcal/mol, are summarized in Table 5. High E(2) values indicate strong conjugation between donor and acceptor components.
Given that the VILD molecule contains relatively few multiple bonds, no significant π→π* interactions were observed, with these interactions exhibiting stabilization energies below 5 kcal/mol. Instead, the molecule is characterized by the presence of five heteroatoms possessing lone pairs, all of which contribute notably to its stability through interactions with neighboring antibonding orbitals. The most substantial stabilization energy (60.7 kcal/mol) originates from the interaction between the lone pair on the N3 atom and the π-antibonding orbital of the O1–C5 carbonyl group.
Furthermore, strong hyperconjugative interactions were identified between the lone pair on the O1 atom and the σ-antibonding orbitals of both the N3-C5 bond in the amide group and the C5–C13 bond. An additional interaction, between the nonbonding electrons of the N6 atom and the σ-antibonding orbital of the C16-C21 bond, contributes over 10 kcal/mol to the stabilization of the molecule.
Table 5 Second order perturbation theory analysis of Fock matrix in NBO for vildagliptin moleculeNatural population analysis (NPA)Natural Population Analysis (NPA), a fundamental component of the NBO methodology, offers a chemically intuitive approach for quantifying atomic charge distributions in molecules. By partitioning electron density into natural atomic orbitals (NAOs), NPA assigns atomic charges that reflect localized bonding patterns, hybridization, and resonance effects, providing a coherent picture of the electronic structure that aligns with classical chemical concepts. This approach rigorously accounts for electron delocalization, including hyperconjugation and lone pair interactions, which is essential for understanding charge-transfer phenomena in bioactive compounds. Developed as an alternative to conventional Mulliken population analysis, NPA exhibits enhanced numerical stability and more accurately describes electron distribution in complex molecules [66].
In the vildagliptin molecule, the atomic partial charges show significant variation (Table 6). The most positive charge (0.7099) is found on the carbon atom C5, which forms part of the carbonyl group. Conversely, the most negative charge (−0.7530) is located on oxygen atom O2, a constituent of the alcohol group attached to the adamantane ring. This markedly negative value is attributed to the substantial electronegativity difference between oxygen and its neighboring atoms. All heteroatoms carry negative partial charges, ranging from − 0.3223 to −0.7530, suggesting that these sites are likely to undergo electrophilic attack.
Most carbon atoms in VILD possess a negative partial charge, with the exceptions of atoms C5, C9, C15, and C21. Notably, C5 exhibits the highest positive charge (0.7099), indicating its susceptibility to nucleophilic attack. This elevated positive charge arises from its double bond with a highly electronegative oxygen atom, as well as its linkage to a nitrogen atom. The nitrogen atoms display partial charges of −0.5119 for N3, −0.6974 for N4, and − 0.3223 for N6. The higher negative charge on N4 is consistent with its single bond to carbon atoms, while N3, which is involved in an amide bond that partially attains double bond character, shows a bit lower negative charge. The lowest negative atomic charge is observed on N6, due to its triple bond with carbon C21.
For hydrogen atoms bonded to carbon, the majority of partial charges lie between 0.19 and 0.22, with the lowest value (0.1899) recorded for hydrogen H29, a member of the pyrrolidine ring. In contrast, other hydrogen atoms exhibit higher charges, such as H23 (0.4571) in the alcohol group, H37 (0.2626) located near the nitrogen atoms of the amide and nitrile groups, and H24 (0.3819) which is directly bonded to nitrogen N4.
Table 6 Natural charges of vildagliptin atomsLocal reactivity descriptorsFukui functions serve as critical descriptors for identifying the most reactive atomic sites within a molecule. By analyzing the electron density distributions of the neutral species as well as its forms with added and removed electron, it is possible to determine whether a given fragment exhibits electrophilic or nucleophilic character. These functions quantify changes in electron density induced by variations in electron count and are mathematically defined as:
$$f\left( r \right)=}}} \right)_}$$
(1)
where ρ(r) represents electron density, N denotes the total number of electrons, and v(r) corresponds to the external nuclear potential.
The function describing electron acquisition (nucleophilic reactivity), denoted as f+(r), is expressed as:
$$\left( r \right)=}}} \right)^+}_}=}\left( r \right) - \left( r \right)$$
(2)
Conversely, the function for electron donation (electrophilic reactivity), f−(r), is defined as:
$$\left( r \right)=}}} \right)^ - }_}=\left( r \right) - }\left( r \right)$$
(3)
Here, r specifies the atomic site, N is the electron count in the neutral state, while ρN+1(r) and ρN−1(r) represent electron densities for the species with added and removed electron, respectively [67].
A refined extension of the Fukui functions is the dual descriptor, which offers better efficiency in characterizing atomic site reactivity. This index is formulated as the difference between f+(r) and f−(r). Positive values of the dual descriptor highlight regions susceptible to nucleophilic attack, whereas negative values indicate sites prone to electrophilic interactions [68].
The Fukui functions were calculated based on natural population analysis, and the values are shown in Table 7. The analysis of the computed Fukui functions for the vildagliptin molecule suggests that a nucleophilic attack is most likely at atom C21, the carbon of the nitrile group. In contrast, markedly lower function values are observed for atom C15 (the adamantane carbon connected to the amino-group nitrogen) and for atom N3, the nitrogen of the amide group within the pyrrolidine ring. Additionally, atoms C17 and C5 exhibit positive f+ values.
All carbon atoms display a propensity for electrophilic attack, except for atom C18, which is situated near an alcohol group. The highest f− values are found at carbon atom C13 of the glycine fragment bonded to an amino group. Moreover, both atom C5 of the amide group and atom C21 of the nitrile group show elevated Fukui function values, while atoms C15 and C10 exhibit a somewhat reduced susceptibility to electrophilic attack.
For radical attacks, atoms C21 and C13 appear to be the most favorable sites, with lower f0 values calculated for atoms C5 and C15. Meanwhile, the dual descriptor highlights atom N4 (a secondary amine) as preferentially prone to nucleophilic attack, likely due to the nearby amide group - a potent electron-withdrawing system. Conversely, the carbon atom C13 bonded to N4 shows the highest tendency toward electrophilic attack.
Table 7 Values of the Fukui indices calculated from natural population analysisGlobal reactivityAn evaluation of frontier orbitals (HOMO - Highest Occupied Molecular Orbital and LUMO - Lowest Unoccupied Molecular Orbital) together with reactivity parameters is fundamental in contemporary computational chemistry, enabling the prediction of reaction mechanisms and the assessment of molecular stability. Density functional theory (DFT) methods offer a quantitative framework for characterizing these properties. In particular, they allow the determination of parameters such as hardness (η) and chemical softness (S), which reflect a molecule’s susceptibility to structural deformation under external charge perturbations. Molecules that are more reactive in interactions driven by polarization are classified as soft, while those resisting charge-induced distortion are regarded as hard. Notably, molecular hardness is enhanced by higher electronegativity, a measure of an atom’s tendency to attract electrons, and by an increased HOMO-LUMO energy gap. Electronegativity (χ) measures an atom’s attraction to electrons, while chemical potential (µ) reflects the likelihood of an electron leaving its orbital. The electrophilicity index (ω) indicates a molecule’s tendency to accept electrons in reactions [69, 70].
The HOMO-LUMO energy gap (ΔE = ELUMO - EHOMO) for the vildagliptin molecule, optimized in vacuum, was determined to be 5.4691 eV (Table 8). This value indicates moderate chemical stability, which is typical for many organic compounds. For context, compounds with significantly higher energy gaps, such as 9.848 eV [71], are generally considered more chemically stable, while those with lower gaps, for example 3.573 eV [72], tend to be more reactive. The high ionization potential (IP = 6.21 eV), representing the energy required to remove an electron from the molecule, points to a low propensity for ionization and, consequently, high chemical robustness. Although the electron affinity is relatively low (0.74 eV), suggesting that only a modest amount of energy is released upon the theoretical formation of an anion, the fact that it remains positive, together with the negative chemical potential (-3.4 eV) and a comparatively high electrophilicity index (2.2), implies a notable tendency for electron acceptance, thereby classifying the molecule as a good electrophile. This characterization is further reinforced by the electronegativity value (3.47), which reflects the molecule’s ability to attract electrons.
Vildagliptin is characterized by a high chemical hardness (2.73) and low chemical softness (0.37), suggesting that it is more predisposed to engage in ion-forming reactions rather than in processes that lead to the formation of covalent bonds.
The molecular orbital model for the HOMO and LUMO is presented in Fig. 7. It can be observed that the electron density in the highest occupied molecular orbital is predominantly localized on the nitrogen atom of the amino group, with partial extension onto carbon atom C13 of the glycine fragment and the carbons C15 and C18 of the adamantane moiety. Conversely, the lowest unoccupied molecular orbital is primarily distributed over the pyrrolidine ring, with a lesser contribution from the nitrile group.
Fig. 7
Frontier molecular orbitals of VILD calculated with DFT
Table 8 Calculated global reactivity descriptors of VILDUV-Vis spectroscopic analysisThe UV-Vis spectrum of vildagliptin was acquired in methanol (Fig. 8) using a 0.001% w/v solution. Experimental results demonstrated strong alignment with computational predictions derived from time-dependent density functional theory (TD-DFT) simulations employing the CAM-B3LYP functional, 6-311++G(2d, p) basis set, and Grimme’s D3BJ dispersion correction. Geometrical optimization and spectral modeling were conducted using the integral equation formalism polarizable continuum model (IEF-PCM) to account for solvation effects in methanol.
Excitation energies and oscillator strengths, calculated via TD-DFT, were processed using GaussSum [73], enabling systematic assignment of electronic transitions to specific molecular orbital interactions with quantified percentage contributions. The first ten excited states of VILD are summarized in Table 9. For mixed-character excitations, natural transition orbital (NTO) analysis was employed to visualize dominant electron-hole pair interactions. This method diagonalizes the transition density matrix, distilling complex excitations into spatially localized charge transfer pathways, thereby clarifying the primary contributors to each transition [74].
Fig. 8
Experimental and theoretical UV-Vis spectra of vildagliptin solution in methanol
Table 9 Calculated absorption wavelength, oscillator and rotatory strength and major contributions to excitationThe vildagliptin molecule does not contain chromophores with strong light-absorbing capabilities, necessitating the input of relatively high energy to achieve electronic excitation. The primary chromophores are the lone pairs on the nitrogen and oxygen atoms, along with the π bonds in the carbonyl and nitrile groups. In the first electronic transition, electron transfer from the HOMO to the LUMO dominates, accounting for 51% of the transition, although the oscillator strength for this process is low. This is reflected in the experimental spectrum, which shows only a subtle inflection at approximately 209 nm. It is also important to note that UV-Vis spectra recorded in solution typically exhibit broadened absorption bands. This broadening arises from solute-solvent interactions (hydrogen bonds, van der Waals forces), which can perturb the energy levels of the solute molecules and lead to loss of fine spectral structure.
The hole NTO is predominantly localized near the lone pair of the amine nitrogen in VILD (Fig. 9), whereas the particle NTO is mainly associated with the π* orbital of the carbonyl group. Subsequent transitions in the computational model are closely spaced. Transitions No. 3 and No. 5, occurring at 197.84 nm and 187.59 nm respectively, display high oscillator strengths and involve the HOMO and higher-lying LUMO orbitals. In the experimental spectrum, these wavelengths correspond to a very intense peak, arising from the overlap of multiple electronic transitions.
For these transitions, the hole NTOs exhibit a distribution similar to that of the first transition, which is consistent with the dominant involvement of the HOMO. In transition No. 3, the particle NTO is primarily localized within the amide group, with a lesser contribution from the nitrile group. Conversely, the particle NTO for transition No. 5 extends over the central region of VILD, encompassing the amine and amide nitrogens as well as the carbonyl group. Transition No. 9, observed at 176.77 nm, exhibits the highest oscillator strength; however, this region falls outside the measurable range of the available instrumentation.
Fig. 9
Representation of natural transition orbitals (NTO) of the “particle” and “hole” pairs for the VILD excitations
Analysis of the density of states (DOS) for molecular orbitals provides valuable insights into the energy-dependent distribution of electronic states and enables the determination of the contributions from specific orbitals to the overall electronic characteristics of the system [75]. In Fig. 10, occupied states are depicted in green while unoccupied states are shown in red. A significant energy gap is evident between the ground and excited states, and the states around 6–7 eV are observed to be very closely spaced.
Fig. 10
Density of state spectrum of VILD
Circular dichroism analysisCircular dichroism (CD) is a UV-Vis spectroscopic technique that elucidates the structure of compounds by measuring the differential absorption of right- and left-circularly polarized light. This method is particularly effective for molecules featuring chiral groups adjacent to their chromophores. Enhanced absorption of left-circularly polarized light results in positive Cotton effects, whereas predominance in right-circularly polarized absorption gives rise to negative bands [76]. Experimental CD spectra were obtained using the same methanolic solution (0.001% w/v) as for the UV-Vis analysis. In parallel, theoretical data were generated via time-dependent DFT (TD-DFT) on a geometry optimized in methanol using the CAM-B3LYP/6-311++G(2d, p) basis set with D3BJ dispersion corrections.
Figure 11 displays the CD spectra, where a low-intensity positive band is observed with maxima at 224 nm (experimental) and 213 nm (computational), alongside a more intense negative band with peaks at 204 nm (experimental) and 195 nm (theoretical). The ordering of the positive and negative Cotton effects is consistent across both datasets, although the theoretical model overestimates the excitation wavelength by approximately 11 nm. This shift may result from not accounting for all specific interactions between the solute and solvent molecules (the IEF-P
Comments (0)