
Remember me

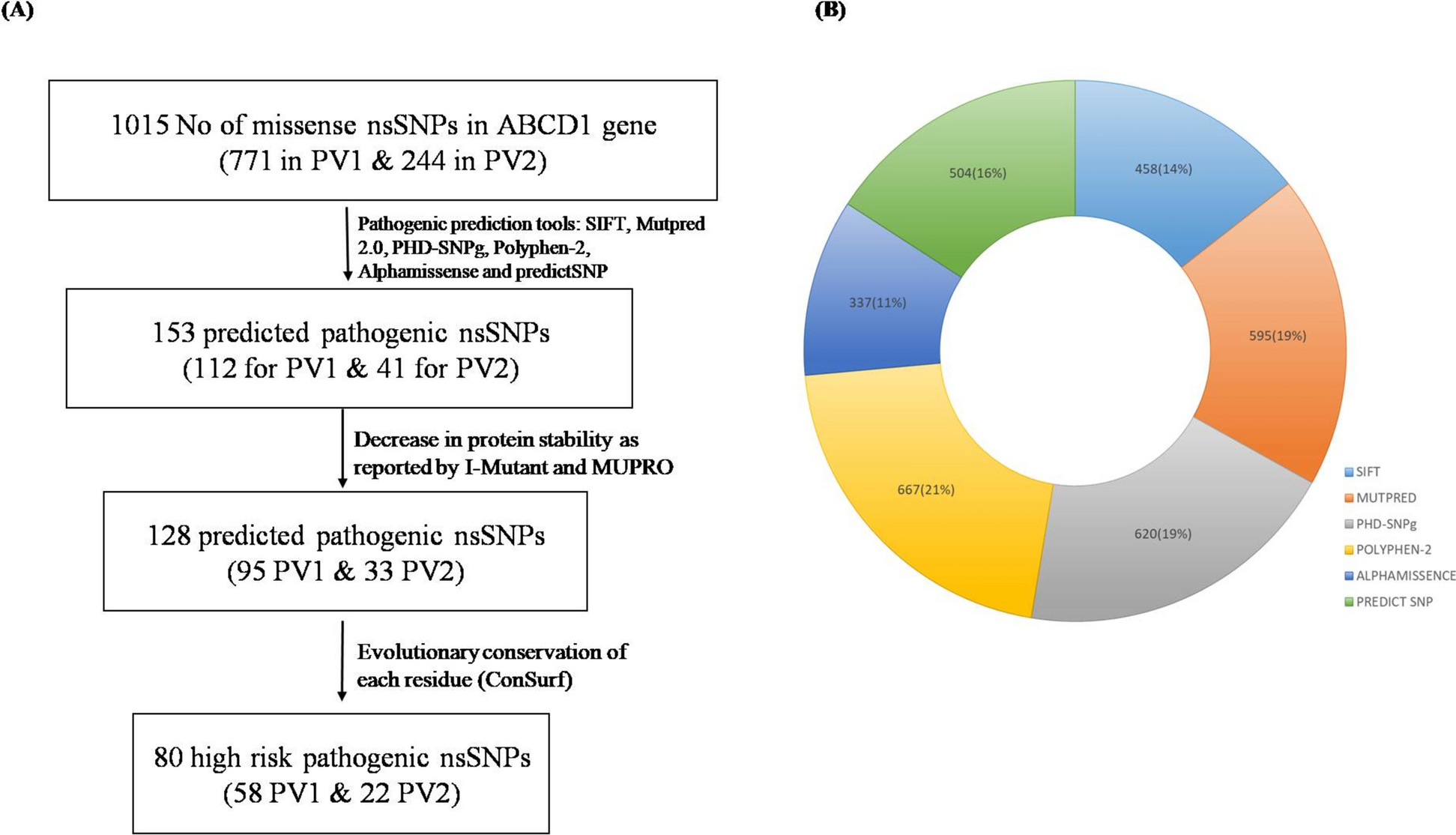

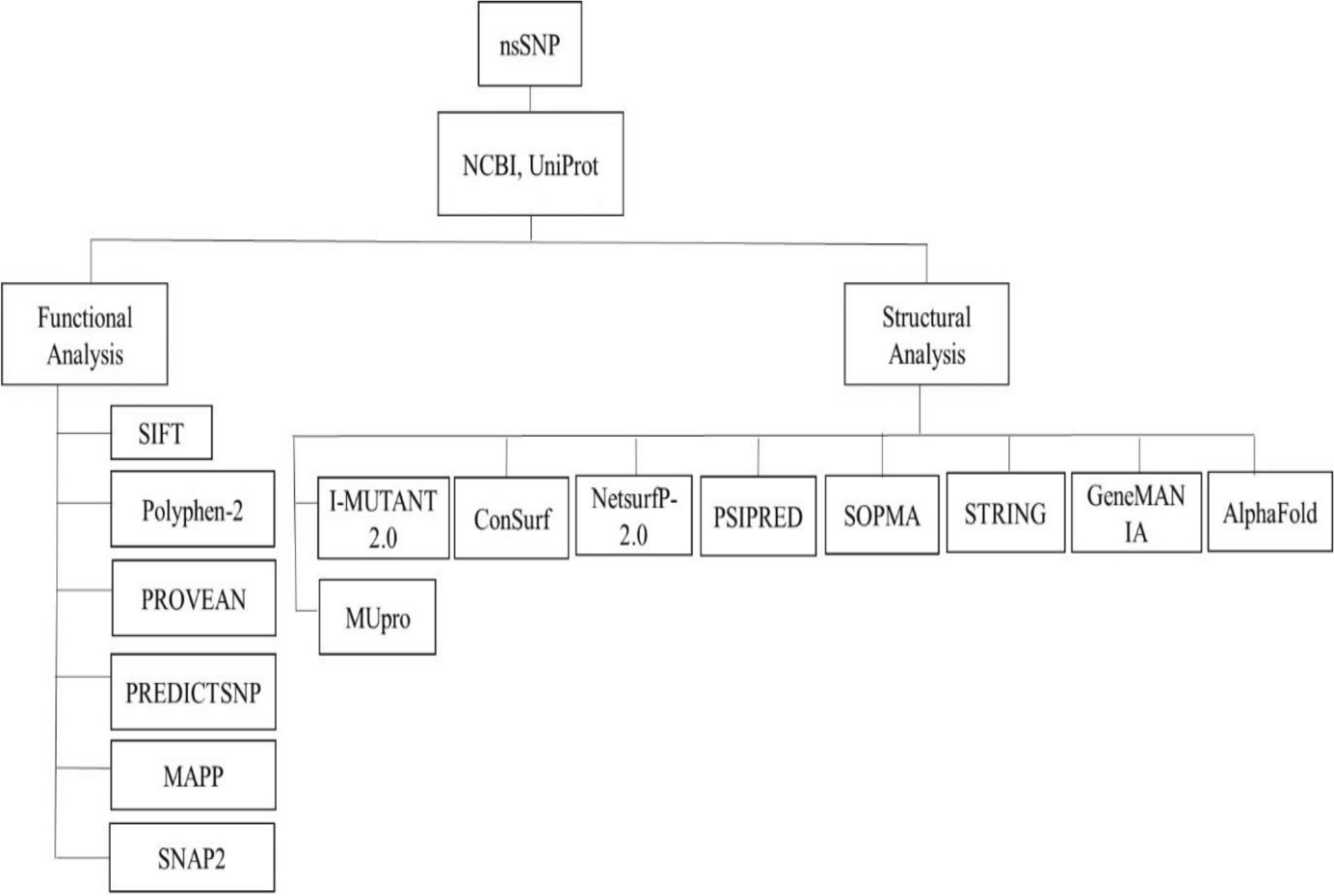

We retrieved SNP data for the ABCD1 gene from the Ensembl database. In total, there were 1015 missense mutations, that occurred due to single nucleotide polymorphism, reported for ABCD1 gene. The most common variant reported for ABCD1 gene is missense variants (61.47%) followed by frameshift mutations (17.2%) [48]. We, therefore, carried out in-silico analysis to predict the pathogenicity of all these missense mutations. Of a total 1015 nsSNPs, 771 were from PV1 and 244 from PV2 transcripts (Fig. 1A). These nsSNPs were subsequently analyzed for their potential pathogenic effects for each protein variants.
Fig. 1
Flowchart showing pathogenicity perdition analysis. A Six different pathogenicity predictors used to predict the pathogenic nsSNPs followed by further filtration based on protein instability and evolutionary conservation analysis that predict the high-risk pathogenic nsSNPs. B Prediction of pathogenic nsSNPs of total 1015 missense mutations from all six pathogenicity predictors (SIFT, Mutpred 2.0, PHD-SNPg, Polyphen-2, Alphamissense and predictSNP). PV1: Protein Variant 1, PV2 protein Variant 2
Determination of pathogenic nsSNPsTo determine whether these nsSNPs impact the structure and function of the ALD protein, we assessed each missense SNP for its pathogenicity using six distinct and widely used in-silico nsSNP pathogenicity prediction methods (PhD-SNPg, PolyPhen2, SIFT, MutPred2, PredictSNP, and AlphaMissense) known for its accuracy.
For PV1 SIFT reported 320 as pathogenic nsSNPs. Polyphen2 predicted 508 as pathogenic nsSNPs. Further, 452 nsSNPs were predicted to be pathogenic by PhD-SNPg, 366 by PredictSNP, 337 by AlphaMissense, and 481 by MutPred2 (Fig. 1B). For PV2 138 by SIFT, 159 by Polyphen2, 168 by PhD-SNPg, 138 by PredictSNP, and 114 by MutPred2 predicted as a pathogenic nsSNPs (Fig. 1B). AlphaMissense were having predictions for PV1 only, so for PV2 we have results from five tools.
In total, 112 nsSNPs for PV1 and 41 nsSNPs for PV2 were predicted pathogenic by all the pathogenic prediction tools (Fig. 1A, Tables 1, 2). For further analysis, these 153 pathogenic nsSNPs were used.
Table 1 Pathogenic nsSNPs of ABCD1 protein variant 1 predicted by all the six toolsTable 2 Pathogenic nsSNPs of ABCD1 protein variant 2 predicted by all the five toolsPredicting the impact of pathogenic nsSNPs on protein stabilityThe three-dimensional structure of proteins is fundamentally determined by their amino acid sequence, which is intricately linked to their functional properties. Variations in the amino acid sequence can influence protein folding, stability, interactions with other molecules, and overall functionality. While mutations at specific locations, such as conserved residues, can significantly alter both the structure and function of a protein however, not all mutations will necessarily affect the protein function.
In this study, we utilized I-Mutant2.0 and Mupro to predict the impact of single-site amino acid mutations on the ALD protein. I-Mutant2.0 estimated that 98 out of 112 non-synonymous SNPs (nsSNPs) of PV1 would decrease the protein’s free energy, suggesting a decrease in protein stability (Fig. 2A). Mupro predicted that 110 out of 112 mutations of PV1 would lead to decreased protein stability. For PV2 out of 41 nsSNPs, I-Mutant2.0 predicted that 33 nsSNPs would decrease the protein stability and Mupro predicted that 30 of these mutations would lead to decrease in protein stability (Fig. 2B).
Fig. 2
Change in protein free energy caused by pathogenic nsSNPs in protein variants of ABCD1 gene as predicted by I-Mutant 2.0 and Mupro. A Change in protein free energy caused by pathogenic nsSNPs in PV1 and B in PV2 of ABCD1 gene. PV1: Protein Variant 1, PV2 protein Variant 2
By integrating the results from both tools, our analysis indicated that 95 pathogenic nsSNPs of PV1 (Table 3) and 33 pathogenic nsSNPs of PV2 (Table 4) (total 128 pathogenic nsSNPs) would decrease the stability of respective protein variants as characterized by a ΔΔG < 0.
Table 3 Impact of pathogenic nsSNPs on protein stability of ABCD1 protein variant 1Table 4 Impact of pathogenic nsSNPs on protein stability of ABCD1 protein variant 2Missense mutation in ABCD1 gene has been demonstrated to causes protein instability and mislocalization, leading to loss of function and absence of β-oxidation activity in ABCD1-deficient fibroblasts [51].
Our analysis showed that 95 pathogenic nsSNPs of PV1 including R660W and 33 pathogenic nsSNPs of PV2 decreased the protein stability (Fig. 1, Table 3,4). Morita et al. (2019) reported that the R660W mutant ABCD1 protein is highly unstable and degraded by non-proteasomal pathways. The protein did not respond to proteasome inhibition or low-temperature rescue. It also failed to restore VLCFA β-oxidation, likely due to mislocalization. These findings supported the computational prediction of decreasing the protein stability and pathogenic phenotype of R660W nsSNP.
Substitution in structured regions (helix, beta sheets) and evolutionary conserved wild-type residues were more likely to result in deleterious phenotype (Khan et al., 2007). To check this, we carried out secondary structure and evolutionary conservation analysis of these 128 pathogenic nsSNPs.
Secondary structure and Evolutionary conservation analysisSecondary structure analysis of ABCD1 protein variantsSecondary structure of PV1 has been derived from the experimentally solved tertiary structure (PDB ID: 7VR1) using DSSP (Fig. 3A, Additional Table 1) [32]. Deriving a secondary structure from an experimentally solved structure using DSSP is generally more accurate than predicting it from the amino acid sequence. DSSP directly analyzes the 3D structure, and thus, provides a precise and detailed picture of secondary structure elements. While prediction methods have improved, they still rely on statistical patterns and can be limited by the complexity of protein folding. We used PDB structure “7VR1” of human ABCD1 PV1 among others, as it has minimum unmodeled small regions and a better resolution of 3.40 Å.
Fig. 3
Protein secondary structure and evolutionary conservation analysis of ALDP. A Secondary structure of ALDP has been derived from experimentally solved structure (7VR1) using DSSP. B Evolutionary conservation prediction of each amino acid residue of ALDP by ConSurf software. The color coding indicated the magnitude of conservation with shades of pink represented evolutionary conserved residues (score 7–9). The letter “e, b, f, and s” represented the ‘exposed’, ‘buried’, ‘highly conserved and exposed,’ and ‘highly conserved and buried residues,’ respectively. The sequence underlined by blue and red solid line indicated the TMD and NBD domain, respectively. The black boxes represented all the wild-type residues that would be affected by HRP_nsSNPs. HRP: High-risk pathogenic
PV1 of ABCD1 was composed of 745 amino acids, and for 598 residues, secondary structure information has been derived. The remaining residues (147 aa) corresponded to the unmodeled regions: 1–67, 438–460, 567–573, and 696–745. Of 598 resides of PV1, 348 (58%) were in alpha helix, 10 (2%) in pi helix, 42 (7%) hydrogen bonded turn, 61(10%) bend, 21(4%) extended strand, 6(1%) in beta bridge, 11(2%) were in 310 helix, 95 (16%) loops and irregular stretches, and 4 aa (1%) participated in polyproline II helix (Fig. 3A, Additional Table 1).
PV2 of ALDP was composed of 227 amino acids. Pairwise sequence alignment using blastp of PV1 and PV2 showed that PV2 significantly aligns (E value: 1e-121, Identity 100%) with TMD domain of PV1. PV2 lacks the 1–185 N-terminal region and aligned to 186–360 region of PV1 (Fig. 4A). The remaining 52 amino acids of PV2 showed poor alignment with PV1. PV2 secondary structure was predicted by SOPMA server, and it consisted of following secondary structures: 161 amino acids (71%) were in alpha helix conformation, 16 (7%) were in extended strand, 47 (21%) amino acids were in random coil, and 3 (1%) were in beta turn (Fig. 4B).
Fig. 4
Pairwise alignment, protein evolutionary conservation, and secondary structure analysis of PV2 of ABCD1 gene. A Pairwise alignment of PV1 and PV2. PV2 aligns to the region 186–360 of PV1, partially covering the TMD domain. The 52aa C-terminal region showed insignificant alignment with PV2. B Evolutionary conservation prediction of each amino acid residue of PV2 by ConSurf software. The color coding indicated the magnitude of conservation with shades of pink represented evolutionary conserved residues (score 7–9). The letter “e, b, f, and s” represented the ‘exposed’, ‘buried’, ‘highly conserved and exposed,’ and ‘highly conserved and buried residues,’ respectively. C Secondary structure prediction of PV2 by SOPMA software. The letter “h, e, c, and t” represents the alpha helix, extended strand, coil and beta turn, respectively. The black boxes represented all the wild-type residues that would be affected by HRP_nsSNPs. PV1: Protein Variant 1, PV2 protein Variant 2, HRP: High-risk pathogenic
We filtered the pathogenic nsSNPs based on evolutionary conservation to get the high-risk pathogenic nsSNPs (HRP_nsSNPs). The proportion of these HRP_nsSNPs in different secondary structures of ALDP was discussed in next section.
Evolutionary conservation analysis of ABCD1 protein variants and localization of high-risk pathogenic nsSNPsThe degree of evolutionary conservation at each residue in the ALD protein was assessed using the ConSurf web server. Residue conservation scores ranged from 1 to 9, with a score of 9 indicating highest conservation. The ConSurf analysis of ‘69 wild-type amino acids that can be replaced by 95 pathogenic nsSNPs in PV1’, identified 62 aa (90%) as evolutionarily significant (score 7–9) and the remaining 7 (10%) as non-significant (score < = 6). Among evolutionarily significant 62 residues, 14 (23%) have a conservation score of 7, and 7 (11%) residues have a conservation score of 8; however, the majority 41 aa (66%) have a conservation score of 9 (Fig. 3B, Table 3).
Consurf analysis of PV2 indicated that of 22 wild-type amino acids that can be replaced by 33 pathogenic nsSNPs, 21 were evolutionarily conserved with 8 aa (38%) having a conservation score of 8, and the rest 13 aa (62%) scoring 9 (Fig. 4C, Table 4).
Next, we classified HRP_nsSNPs among these 128 pathogenic nsSNPs (95 PV1 and 33 PV2) based on the following criteria: (1) nsSNPs were predicted pathogenic by all pathogenic prediction tools. (2) The pathogenic nsSNPs were predicted to decrease the stability of the protein by both I-Mutant2.0 and Mupro tools. (3) When such pathogenic nsSNPs (mentioned in the above two points) were positionally highly conserved (score 9). These HRP_nsSNPs were expected to have a significant impact on protein function and structural properties. This gave us 80 HRP_nsSNPs (58 PV1 & 22 PV2) (Fig. 1A, Tables 5, 6).
Table 5 High-risk pathogenic nsSNPs of ABCD1 protein variant 1Table 6 High-risk pathogenic nsSNPs of ABCD1 protein variant 2The 58 HRP_nsSNPs of PV1 corresponds to 41 wild-type amino acids (wt_aa) that can be replaced by 58 HRP_nsSNPs. Almost wild-type residues, which would be affected by HRPnsSNPs, all were located in the conserved domains (TMD (61%) and NBD (34%)) except two residues (5%): A398T and V419M (Fig. 3B, Table 5). These were located in the inter-domain linker region. As PV2 corresponded to the TMD domain, thus, all HRP nsSNPs of PV2 were located in TMD domain only (Table 6). Additionally, the secondary structure analysis of HRP_nsSNPs of PV1 (n = 41 wt_aa) showed, 23 aa (56%) were located in alpha helix, 3 (7.3%) in pi helix, 2 (5%) in extended strand, 7 (17%) in bend, 3 (7.3%) in hydrogen bonded turn, and 3 (7.3%) were located in loops and irregular stretches (Table 5).
Further, 22 HRP_nsSNPs of PV2 corresponds to 13 wild-type amino acids (wt_aa) that can be replaced by 22 HRP_nsSNPs. The secondary structure analysis highlighted that 9 aa (69%) were located in alpha helix, 1 (8%) in extended strand, 2 (15%) in random coil, and 1 (8%) in beta turn (Fig. 4B).
In both the PVs, majority of the HRP_nsSNPs occurred in α helices (56% and 69%, respectively). Our results were in line with those published previously for disease-causing mutations in protein secondary structures. Comprehensive study that analyzed 1939 missense mutations in secondary structures to get insights about the spectrum of disease-causing mutations in protein secondary structures revealed that majority (48%) of the disease‑causing mutations occurred in α helices, however, β‑strands accounted for lesser proportion (28%) [38].
Further, mapping the predicted HRP_nsSNPs to the genomic location would highlight the mutation hotspots. HRP_nsSNPs of PV1 localized to several exons including exon 1 (57%), followed by exon 6 (16%), exon 9 (12%), and exon 2 (5%). Exons 7, 8, 3, and 4 collectively accounted for the remaining 10% (Table 5). Two exons (1 and 2) corresponded to the cDNA of PV2 and almost all its the HRP_nsSNPs corresponded to exon1 (91%). Like PV1, exon 2 accounted for relatively lower (9%) pathogenic nsSNPs (Table 6). Our results corroborated well with the mutation hotspots study of ABCD1 where the hotspots of missense variants have been reported within exons 1, 2, 6, 8, and 9 [10]. This alignment underscores the relevance of these exons as critical regions for genetic variation in the ABCD1 gene.
In the context of protein, of all the HRP_nsSNPs of PV1 predicted in our study, 12 (R104H, I151T, S213Y, S213F, K217N, P218L, T254A, T254M, L258P, Y337C, G343R, and G343D) were located in all the 6 conserved transmembrane segments (TS) except TS5 (Table 5). Among these 12, six (S213Y, S213F, K217N and P218L of TS3 and Y337C, and G343R of TS6) has not been reported earlier and were not identified in any of the ALD case. In PV2, 3 HRP_nsSNPs (P33L, D36N, and D36A) were located in TS3 and none were reported earlier for any of the ALD cases.
Collectively, we found that the majority of the HRP_nsSNPs occurred in structured regions (helix, beta sheets) instead of unstructured regions, mostly occupied the hotspots of missense variants suggesting that these HRP_nsSNPs were more likely to affect protein stability, its interacting partners and protein folding. These were analyzed in the next section.
Analysis of solvent accessibility & physical and chemical properties of ALD proteinThe solvent accessible surface area was examined using the web program GetArea to identify the regions where mutations were more likely to have harmful effects on protein structure and function. Of 41 wild-type amino acids (wt_aa) that can be replaced by 58 HRP_nsSNPs in PV1, 22 (54%) were predicted to be buried, 9 (22%) to be solvent exposed, and the rest 10 aa (24%) were predicted to be located in different regions of the proteins (Fig. 5A, Table 5). Of the 13 wild-type amino acids that can be replaced by 22 HRP_nsSNPs in PV2, 2 (15%) were predicted to be buried, 4 (31%) to be solvent exposed and the rest 7aa (54%) were located in other regions of the protein (Fig. 5B, Table 6). This suggested that mutations in either core or solvent-exposed region of the protein were more likely to have deleterious effect on structure and function of the protein probably by destabilizing the protein folding or disrupting the protein interactions.
Fig. 5
Amino acid residues predicted for being solvent exposed or buried by GETAREA. The solvent accessibility was predicted for A All the HRP_nsSPNs of PV1 and B PV2. PV1: Protein Variant 1, PV2 protein Variant 2
To get insights into the physico-chemical impacts of HRP_nsSNPs, we carried out HOPE analysis. Results showed that all 58 HRP_nsSNPs of PV1 brought about changes in the size of amino acids. Majority, 32 (55%) wild-type amino acids were substituted by large-sized amino acids, rest 26 (45%) were substituted by small-sized amino acids. Twenty-six HRP_nsSNPs (45%) resulted in a change of charge. Additionally, 47 (81%) HRP_nsSNPs had an impact on the hydrophobicity of the protein, 20 HRP_nsSNPs decreased the hydrophobicity, however, 27 HRP_nsSNPs increased the hydrophobicity (Table 7).
Table 7 Physical and chemical properties of ALD protein and structural effects of HRP_nsSNPs on protein variant 1The HRP_nsSNPs: G116R, G343D, G343R, G510D, G512R, G512S, G512V, and G529D, exhibit glycine as their wild-type residues that enabled flexibility for torsion angles. HOPE predicted that the unusual torsion angles caused by substituted residues led to force the local backbone into an incorrect conformation and disrupt the local structure. Our study confirmed the pathogenicity of the known G512S mutation in the conserved Walker A motif “GPNGCGKS” (position: 507–514), which is essential for ATP binding in ALDP [67]. This mutation had been reported as pathogenic and identified in 49 ALD cases [33, 43, 57, 60, 77]. It had been linked to reduced protein levels and ATPase activity in ALD patients [67]. Importantly, our study not only captured such pathogenic missense mutation “G512S”, rather predicted two novel variants being HRP_nsSNPs “G512R and G512V” (Table 7). These findings extended the existing knowledge and further emphasized the structural and functional sensitivity of this critical motif.
For 26 HRP_nsSNPs: A294T, D200N, E291D, E291K, E292K, F130C, G510D, H659Y, K217N, L313Q, N148S, N148Y, P263S, Q544R, R104H, R163C, R163G, R163H, R163L, R163P, R189W, R518G, R554S, R660Q, R660W, and T254A, HOPE predicted the loss of interactions like: hydrophobic, external interactions, ionic interaction with other molecules, and other part of the protein.
Wild-type residue of HRP_nsSNPs E291K, E291D, E292K, and A294T, were the crucial residues of the EAA-like motif “NSEEIAFYGGHEVEL” (position: 289–303) which was reported important for substrate specificity in certain eukaryotic ABC transporters [71, 74]. Previous studies of ALD patients reported disease-causing mutations in its conserved glutamic acid residues [14, 42, 83]. Our computational analysis further supported these findings, identified substitutions within this motif E291K, E291D, E292K, and A294T, as high-risk pathogenic. This showed its critical role in ALDP function. Further, these HRP_nsSNPS were predicted to likely disrupt the multimeric interactions, either with other molecules or with different regions of the protein as per HOPE analysis. This suggested that these residues might contribute to the functional specificity of the transporter by influencing its structural or interaction dynamics.
Our study aligned well with earlier findings on the functional importance of the 22 residue long loop1 motif “LVSRTFLSVYVARLDGRLARCI” (position 101–122) of ALDP. Previous reports showed that mutations in conserved residues of this motif, e.g., agrinine (R) at position 104 and glycine (G) at position 116 were linked to ALD as reported in > 15 ALD patients [42]. Our computational analysis predicted R104H and G116R as high-risk pathogenic variants. Additionally, mutation G116R was known to disrupt protein structure and interactions, including ALDP-ATP-citrate lyase (ACLY) interaction, leading to disease, this supported the HOPE prediction of disrupting the local structure [28]. This highlighted the relevance of our findings in the context to established disease mechanisms.
These HRP_nsSNPs: N148Y, R189W, T198M, S213F, S213Y, T254M, P263S, A294T, L313Q, R518G, R554S, Q611R, S656F, S656P, T658I, H659Y, R660Q, and R660W, were predicted to show the effect of change in hydrophobicity resulting in loss of hydrophobic interaction either in core of the protein or with other molecules and affected the hydrogen bond formation in neighboring residues. HRP_nsSNP ‘Q611R’ indicated the pathogenic potential of Q611R mutation in ABC signature motif “LSGGEKQRIGMARMF” (position: 605–619) of ALDP, a region essential for ATP binding and hydrolysis [44, 48]. This variant predicted to alter the hydrophobicity and disrupt the key hydrogen bonds thus, potentially impairing the protein function. Notably, Q611R has been reported in 2 ALD cases and identified in ALD newborn screening supporting its clinical relevance [63].
Additionally, the following HRP_nsSNPs G343D, G343R, H659Y, S656F, S656P, T658I, and Y174C, were predicted to disrupt the protein folding. Overall, HOPE results imply that modifications in the physico-chemical characteristics, resulting from amino acid substitution at these locations, significantly alter the amino acid properties of the ALDP. These alterations were likely to disrupt the inter- and intramolecular interactions of ALDP, potentially impacting its structure and function.
Gene–gene interaction networkGene–gene interaction network of the ABCD1 gene predicted by GeneMANIA showed that it physically interacts with ABCD2, ABCD3, PEX19, PEX3, and ACADS along with other proteins as well, as shown in Fig. 6. ABCD1 protein is a peroxisome membrane transport proteins required for the import of coenzyme A-activated VLCFAs into peroxisomes for β-oxidation [56, 79]. Like ABCD1, ABCD2 also transports VLCFAs into peroxisomes, whereas, ABCD3 protein functions in the transport of bile acid intermediates into peroxisomes [34]. PEX19, a chaperone protein, binds to peroxisomal membrane proteins and targets them to the peroxisome membrane [69], whereas, PEX3, a docking protein, interacts with PEX19 and other proteins to facilitate them to embed into the peroxisome membrane [23]. ACADS, a short-chain acyl-CoA dehydrogenase, catalyze the first step of mitochondrial fatty acid beta-oxidation pathway [78]. This indicates that HRP_nsSNPs of the ABCD1 gene may affect the interaction and functioning of other genes in the gene–gene interaction network.
Fig. 6
ABCD1 gene–gene interaction network predicted by GeneMANIA. Gene–gene interaction network of the ABCD1 gene shows networks of the gene based on physical interactions (red), co-expression (purple), predicted (orange), co-localization (blue), genetic interactions (green), pathway (cyan), and shared protein domains (beige). Node size indicated the strength of interaction; however, the edge colors represent the type of relationship. Key genes included in this network were: ABCD2: ATP-binding cassette sub-family D member 2, ABCD3: ATP-binding cassette sub-family D member 3, PEX3: Peroxisomal biogenesis factor 3, PEX19: Peroxisomal biogenesis factor 19, ACOX1: Acyl-CoA oxidase 1, ACADS: Acyl-CoA dehydrogenase short chain, SLC27A2: Solute carrier family 27 member 2, AGPS: Alkylglycerone phosphate synthase, CHCHD3: Coiled-coil-helix-coiled-coil-helix domain, IMMT: Inner membrane mitochondrial protein
nsSNVs of ABCD1 from Indian populationsThe IndiGen Project, a 1000 genome project of India where whole-genome sequencing of diverse ethnic Indian populations (n = 1029) carried out to generate comprehensive resource of genetic variants comprising of single nucleotide variants and indels [30]. For ABCD1 gene, 12 nsSNVs, all from exonic regions, have been identified. While the majority (n = 9) were previously documented and identified as non pathogenic, three nsSNVs: A100T, V250L, and I519V were found to be unique to the Indian population, with low allele frequencies of 0.001 and 0.0005 (Table 8). The analysis of all these three substitutions revealed significant effects on protein structure and stability. All substitutions were predicted to reduce protein stability, as the wild-type residues, originally buried within the protein core, were replaced by larger ones. These changes led to alterations in hydrophobicity while maintaining the charge of the amino acids. These unique genetic variants highlight the importance of population-specific studies in uncovering novel/rare genetic variations. It has been reported that over 18 million unique variants in Indian genomes have been identified through comprehensive analysis of the whole-genome sequencing (WGS) of 1029 healthy Indian individuals [30]. These variants provide a foundation for further studies to explore their potential functional significance, roles in disease susceptibility, and implications for population-specific therapeutic strategies.
Table 8 nsSNVs of ABCD1 from Indigenome database
Comments (0)