AS presents significant diagnostic challenge in its early stages due to its insidious onset and lack of specific clinical manifestations. The complex genotype–phenotype correlations further complicate early recognition. Many patients remain undiagnosed until substantial renal impairment or even renal failure occurs. Hematuria and proteinuria are the most common clinical features across all genetic subtypes of AS, while extrarenal manifestations such as SNHL and ocular abnormalities are closely related to the specific type of collagen IV gene mutation, as well as the patient’s age and sex [4, 5]. Although these extrarenal signs can aid diagnosis, they may be absent or overlooked, particularly in patients with mild disease.
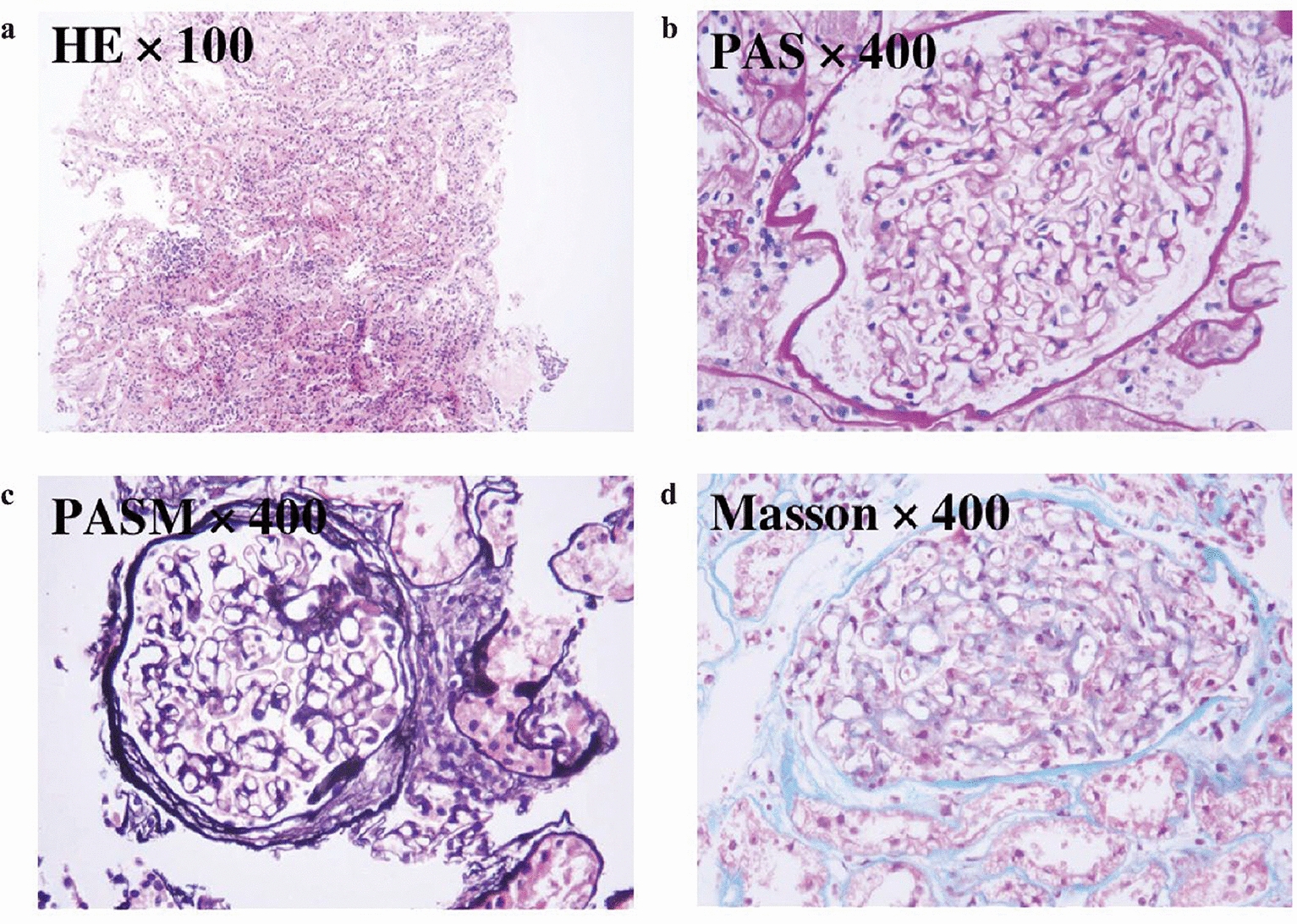
Histologically, AS often lacks distinctive features under light microscopy, and immunofluorescence studies are frequently negative during early stages [6]. Electron microscopy may reveal diffuse thinning, irregular thickening, or segmental wrinkling of the GBM, accompanied by podocyte foot process effacement. However, in some cases, GBM changes may be nonspecific and limited to diffuse thinning [7]. Thus, a definitive diagnosis requires an integrated approach involving clinical features, laboratory tests, renal histopathology, family history, and molecular genetic analysis. With the growing availability of NGS, genetic testing has become one of the most sensitive and accurate diagnostic tools. It not only facilitates timely diagnosis but also offers prognostic insights and valuable guidance for genetic counseling and family screening [8].
ADAS is a frequently underrecognized form of hereditary nephropathy due to its mild, slowly progressive course. Compared with XLAS or ARAS, ADAS typically presents with later onset and rarely features extrarenal involvement. Some patients may exhibit only isolated hematuria or proteinuria throughout their lifetime [9]. Histopathological findings are often nonspecific, with GBM thinning or mild podocyte foot process effacement being the predominant changes. In our case, light microscopy revealed 15 glomeruli, of which 6 (40%) showed global sclerosis, despite the remaining glomeruli demonstrating only mild mesangial hypercellularity and mesangial matrix expansion without segmental sclerosis. This proportion of globally sclerotic glomeruli was higher than might be expected from the otherwise benign appearance of the non-sclerotic glomeruli. This finding is consistent with previous descriptions in ADAS, where chronic, slowly progressive injury can lead to complete sclerosis in a subset of glomeruli while others remain morphologically preserved until later stages. In the absence of a positive family history, these patients are easily misdiagnosed as having primary glomerulopathies, such as IgA nephropathy or nonspecific mesangial proliferative lesions [10].Although the risk of progression to end-stage renal disease (ESRD) is generally lower in ADAS, proteinuria, hypertension, and declining renal function serve as markers of disease progression [2]. Studies have shown that approximately 39% of ADAS patients with proteinuria ultimately require renal replacement therapy, and 10–20% progress to ESRD before the age of 70 [11].
In the present case, NGS identified a heterozygous COL4A4 variant (c.913G > C, p.Gly305Arg), located at chr2:227,967,522 (GRCh37), resulting in the substitution of a conserved glycine residue within the collagenous triple-helical domain, which is crucial for the stability of the type IV collagen network. Glycine residues play a central role in maintaining the tightly packed triple helix; their substitution with bulkier amino acids such as arginine can disrupt the collagen structure, leading to functional impairment of the GBM. This variant has not been previously reported in major population databases such as gnomAD (https://gnomad.broadinstitute.org/) or ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), indicating its rarity. Multiple in silico predictive algorithms suggested pathogenicity. Moreover, similar glycine substitutions in COL4A4 (e.g., p.Gly1018Arg, p.Gly285Ala) have been previously reported in ADAS patients and classified as pathogenic [12, 13]. According to the American College of Medical Genetics and Genomics (ACMG) guidelines [14], this variant can be classified as “likely pathogenic” (criteria: PM1 + PM2 + PP3_Strong). This novel mutation expands the known mutational spectrum of COL4A4. It is worth noting that the proband’s son also carries the same mutation, while her daughter does not. Both children underwent urine testing, and while the son currently shows no obvious symptoms, it is recommended that he undergo regular follow-up due to individual differences.
At present, there is no definitive cure for AS. Early intervention is crucial to delay renal disease progression and preserve renal function. Studies have demonstrated that inhibition of the renin-angiotensin-aldosterone system (RAAS) significantly delays the onset of renal failure, particularly when initiated before a marked decline in glomerular filtration rate [15]. Clinical practice guidelines recommend early initiation of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) in ADAS patients once proteinuria is detected [16]. The EARLY-PROTECT trial is investigating the potential benefits of initiating ACEIs even before the onset of proteinuria [17]. Furthermore, sodium-glucose co-transporter 2 (SGLT2) inhibitors, such as dapagliflozin, have demonstrated efficacy in slowing chronic kidney disease progression and are being actively explored as potential therapies for AS [18]. However, in this case, the confirmation of ADAS through genetic sequencing did not result in any immediate change in the treatment plan. While genetic sequencing is crucial for confirming the diagnosis and understanding the genetic basis of the disease, the management of ADAS typically remains supportive in the absence of clear clinical indications. Treatment such as RAAS inhibition is guided by clinical factors like proteinuria and renal function, and while genetic confirmation is important for long-term management, it may not always lead to immediate modifications in treatment strategy.
In this case, the patient initially presented with hematuria and subsequently developed nephrotic-range proteinuria. For a prolonged period, the patient was managed following the protocol for primary glomerulonephritis. Independently, she chose to receive traditional chinese medicine (TCM) concurrently, but no satisfactory clinical improvement was achieved, and significant psychological distress was noted. After renal biopsy revealed extensive GBM thinning on electron microscopy, genetic testing was pursued, leading to a definitive diagnosis of ADAS. The patient was subsequently advised to discontinue ineffective medications, focus on blood pressure control, and adhere to evidence-based therapies to slow disease progression. Following treatment adjustments and psychological support, her condition stabilized.
This case underscores the importance of considering hereditary nephropathies, such as AS, in patients with hematuria or proteinuria without obvious extrarenal manifestations or a positive family history. When biopsy findings suggest the possibility of a hereditary disorder, clinicians should maintain a high index of suspicion and promptly recommend genetic testing to facilitate early diagnosis and intervention. Advances in targeted therapies and gene editing technologies are anticipated to further refine the management of ADAS in the future.




Comments (0)