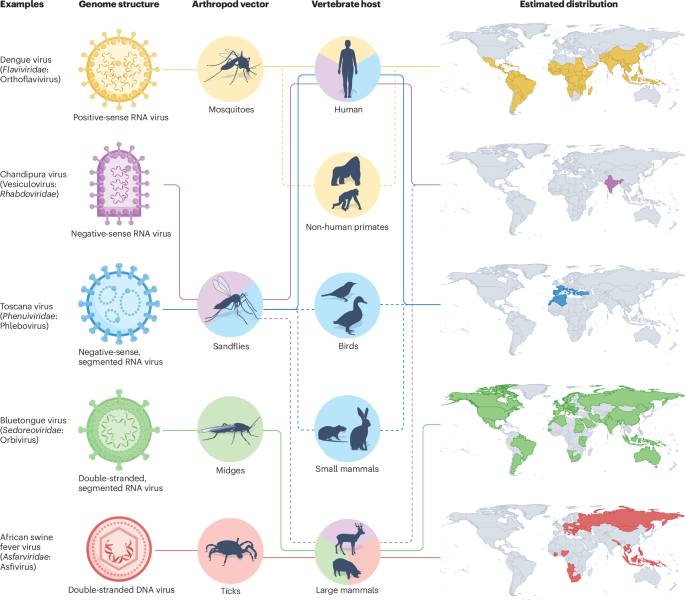
Determining variant effects with pooled prime editing
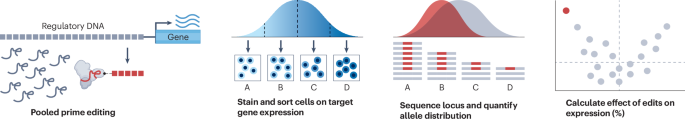
Over one billion genetic variants have been identified in humans. However, most of these variants lack functional characterization and whether they affect human health remains unclear. Multiplexed assays of variant effect (MAVEs) can close this gap by enabling the phenotypic consequences of thousands of genetic variants to be determined in a single experiment. Among MAVEs, assays that use genome editing offer a unique advantage: they test variants in their native context, thereby capturing effects on both gene expression and protein function. Still, editing-based MAVEs are limited by the number and type of variants that can be tested.
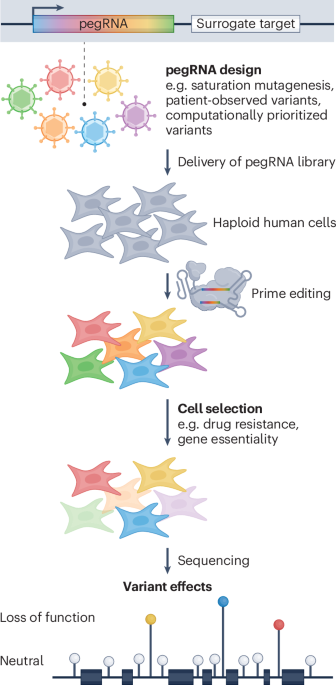
First described by Anzalone et al., prime editing stands out as a potentially powerful solution because it allows almost any short variant anywhere in the human genome to be installed with high precision. Prime editing relies on two main components: the prime editor and the prime editing guide RNA (pegRNA). The prime editor, a fusion of a Cas9 nickase with a reverse transcriptase, writes the desired edit into the genome. The pegRNA both specifies the genomic target site and provides the template for the genetic change. Low editing efficiencies have hampered efforts to use prime editing for variant screening, but recent improvements to the technology have made such screens possible.

Comments (0)