Statistical methods for primary and secondary outcomes
All data will be described using the appropriate descriptive statistical methods, including means, SD, quartiles, minimum and maximum values for continuous variables, and absolute and relative frequencies for categorical variables. Additionally, 95% confidence intervals (95% CI) will be calculated to estimate the unknown population parameters.
Efficacy measures will be analysed using regression analyses based on the general linear or generalized linear model to accommodate the intra-patient structure of correlations intrinsic to repeated measurements over time. These models also allow adjustment for covariates.
We will use the general linear model whenever possible. Normality tests will be performed on the sample data by the Kolmogorov–Smirnov method, the Shapiro–Wilk method or both. If significant results are obtained, alternatives based on the generalized linear model shall be used.
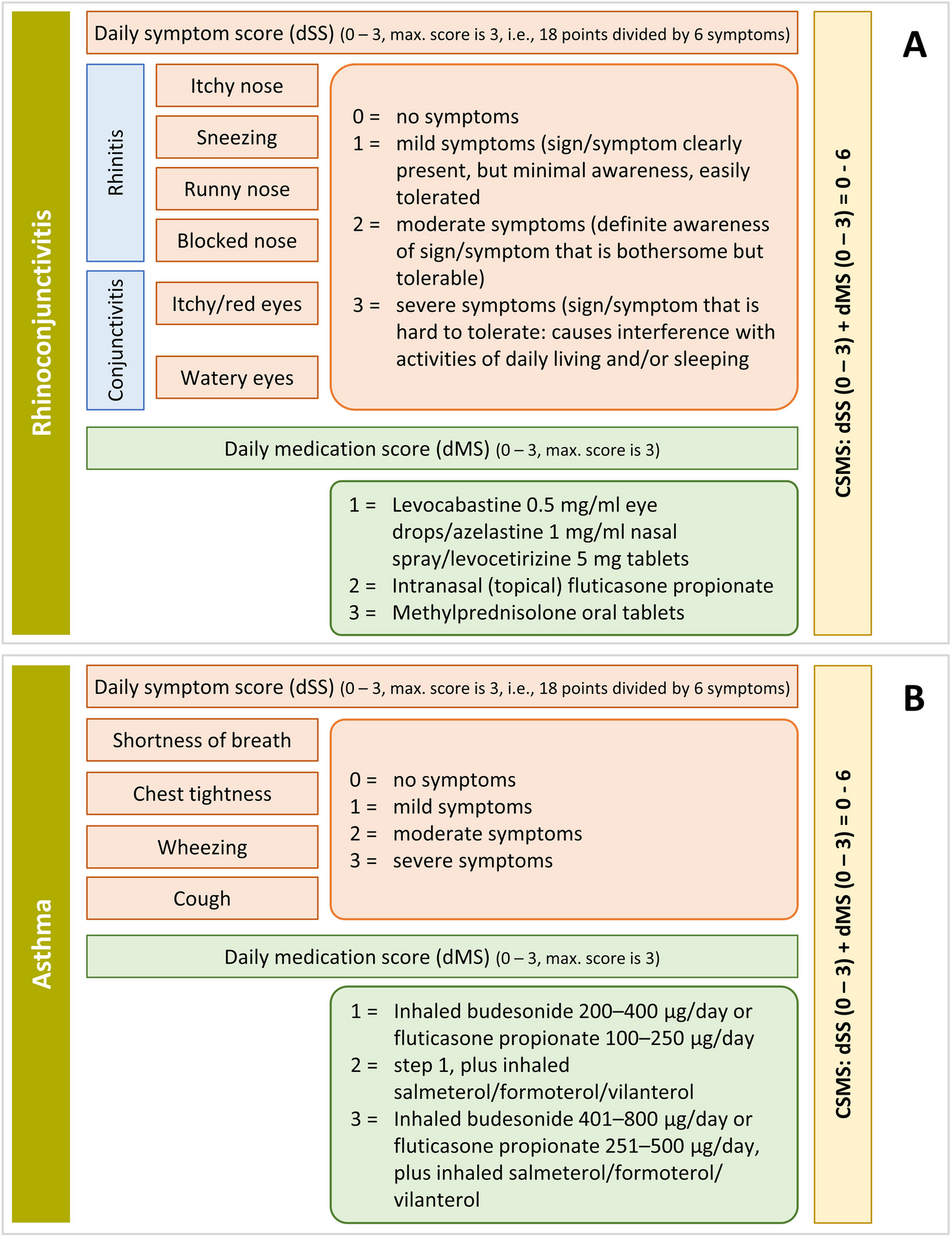
The primary efficacy measure values at each visit will be obtained by averaging the daily values recorded in the participant diaries during the period corresponding to each visit. The primary outcome measure is the CSMS4, as recommended by the EACCI (see the Table 2 for a description). The CSMS4 shall be obtained repeatedly throughout the study. The main hypotheses (null and alternative) are defined as:
CSMS4Exp and CSMS4Pb refer to the scores of the aforementioned four-symptom rhinitis scale in the experimental and placebo groups, respectively, at any post-baseline study visit. Notably, this formulation implies a superiority test of the experimental treatment versus placebo. Such superiority may be tested for any post-baseline visit individually or for the entire scores’ trajectory throughout the study.
The mixed models for repeated measures will include treatment, study visit, and site as fixed factors, baseline as a covariate, and patient as a random (intercept) factor. Other fixed factors may be incorporated that have prognostic value on the primary efficacy measure (which shall be specified in the SAP, if needed). We will use the estimate of the fixed treatment factor and the p-value associated with the Wald-type contrast for the hypothesis tests. The adjusted means for each treatment will also be provided together with their corresponding 95% CI. We will retain the interaction between treatment and visit to obtain the adjusted means per treatment and visit. If the centre factor is significant, we will also try to retain the interaction between treatment and centre (and present the means adjusted by centres) to assess possible differences in the effects between centres.
A logarithmic data transformation will be initially attempted if the normality assumptions are not met. If normality assumptions remain unmet, we will use the generalized linear model with a Gamma distribution for errors and a (natural) logarithmic link functions. In such a case, the adjusted means would be retro-transformed to ease its interpretation.
Several secondary efficacy measures (CSMS, dSS4, dSS6, dMS) are similar to the primary efficacy measure. Hence, they will be analysed following the same procedure. The mini-RQLQ, RCAT, and VAS scores will also be analysed similarly to the primary efficacy measure. We do not expect the RCAT and VAS scores to deviate from normality and intend to use the general linear model; however, the VAS results may require a logarithmic transformation. If the normality remains unmet, we would proceed as described.
Disease-related costs will be calculated as the product of the number of times each healthcare unit resource cost was used by the corresponding resource’s unitary cost (in euros, €); the accumulated costs will be compared at the end of the study. We will only analyse the data from patients who complete cost-related information in all follow-up visits and with at least 9 months of study participation (to avoid distortions caused by missing values due to dropouts), except in case of death. We will perform two unilateral t-tests to test for equivalence with the equivalence thresholds that will be specified in the statistical analysis plan (SAP). In addition, we will calculate a cost-effectiveness measure as the quotient between each patient’s accumulated cost and the quality-adjusted life years (QALY) calculated from the mini-RQLQ. These measures represent the cost of obtaining a unit of effect (i.e. a QALY) with the corresponding treatment. Last, incremental cost-effectiveness ratios will be calculated with respect to the placebo control from the public insurance health perspective to ascertain whether and how the AIT under study affords benefits to the health system.
Safety measures will include the adverse events and laboratory data. Adverse events will be coded by Systems and Organs to the Lowest Level Term according to the Medical Dictionary for Regulatory Activities (MedDRA) and described by severity, seriousness, and relationship to investigational products. In addition, adverse reactions to the investigational products will be further classified as local or systemic according to the World Allergy Organization (WAO) grading system [21].
Safety data will be analysed on a safety analysis population comprising all randomized patients who receive at least one dose of the investigational product. Efficacy analyses will be performed over a modified intention-to-treat population that will be a subset of the safety population comprising patients who have both baseline and at least one post-baseline assessment of the primary outcome measure reported. If more than 10% of randomized patients had relevant protocol deviations, a per-protocol population would be defined to exclude these, and the efficacy analyses would be repeated in this subset to assess the influence of such deviations on the results.
The SAP will be made available and will further detail the statistical analyses. All statistical analyses will be performed using the version 9.4 or higher of the SAS software.
Interim analyses
No interim analyses are expected for this study with the exception of the blinded safety data summaries that will be provided at two prespecified timepoints to the Data Safety Monitoring Board (DSMB, see item 21a). No decision pertaining trial continuation or modification in any way will be taken as a result of the assessments by the DSMB.
Methods for additional analyses (e.g. subgroup analyses)
No subgroup analyses are initially planned. However, the possible influence of the patient’s sex and age and the presence of asthma or other concomitant diseases on the results should be explored by introducing these variables into the regression models provided for the analysis of the efficacy measures. If significant interactions are detected in any of these models, we shall consider performing the corresponding subgroup analyses, but these will be always regarded as exploratory.
Methods in analysis to handle protocol non-adherence and any statistical methods to handle missing data
We will perform sensitivity analyses if more than 20% of the participants have missing CSMS4 values on 20% or more of the days preceding each assessment visit. We will repeat the procedure described before excluding these patients.
Since the previous analyses concern models for repeated measures from each patient, these can accommodate random data missing mechanisms—i.e. not specifically related to the interventions, which constitute a reasonable assumption in this study. In the event of an aberrant pattern of missing values, for example, more than 20% of intermediate values erratically cancelled due to incorrect completion of the participant’s diaries, we will apply imputation techniques using simple linear interpolation before running the analyses.
Suppose relevant between-participants differences are evidenced regarding their exposure times. In that case, we may incorporate the visit factor into the aforementioned models as random instead of a fixed factor, which implies the incorporation of random slopes into the models. In this case, we would give further details in an update of the SAP.
As mentioned, efficacy analyses will be performed following the intention-to-treat ideal using an analysis set (herein called modified intention to treat population, see § 20a) that will include as much of randomized subjects as possible. The only exceptions, to safeguard the integrity of the analyses, will be patients who do not receive any dose of the IMP or do not have any data post randomization. This is considered to yield conservative estimates, since in general protocol non-adherence issues will tend to diminish the estimated treatment effect in a superiority trial like this.
Plans to give access to the full protocol, participant-leveldata and statistical code
The sponsor of the study will oversee the dataset and the data analysis. Granting access to this information will be allowed on a personalized basis, upon request by the interested party, and with permission from the corresponding author of this article, who will oversee the study. Data access requests should be addressed to the email address included in the article header.









Comments (0)