
Remember me
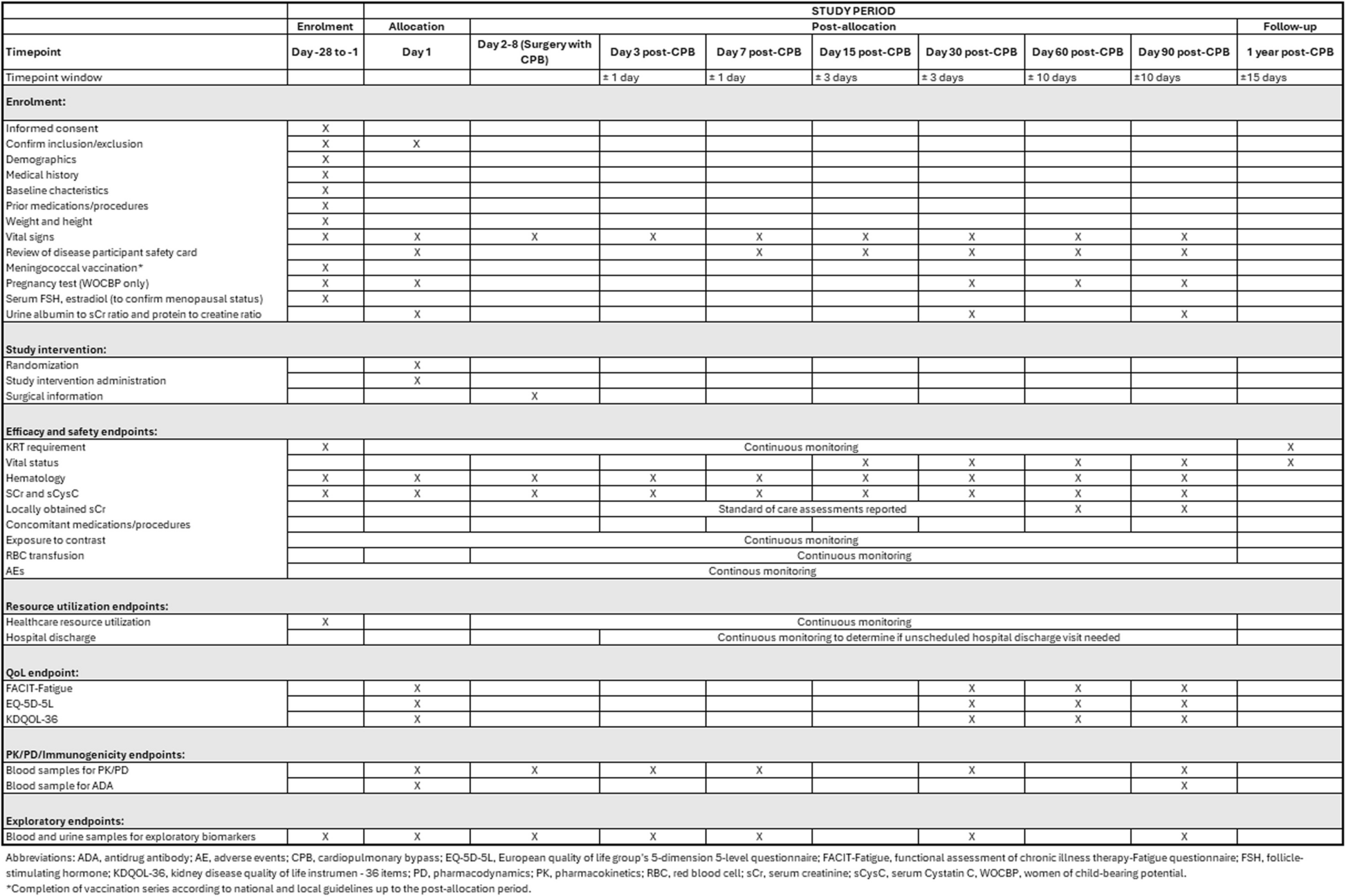








SPIRIT guidelines were followed in the reporting of this trial [35]. The completed SPIRIT checklist can be found in Additional File 1, and the SPIRIT Figure detailing the full study schedule is included as Fig. 1.
Fig. 1
SPIRIT Figure study period schedule
Study designARTEMIS is a phase 3, randomized, double-blind, placebo-controlled, global study of ravulizumab in adult participants with CKD undergoing non-emergent cardiac surgery requiring CPB for coronary artery bypass graft (CABG), valve replacement or repair, or combined procedures. The primary objective of this study is to assess the efficacy of ravulizumab in reducing the risk of MAKE at 90 days following surgery with CPB (MAKE90).
Participants are included on the basis of having preoperative CKD and considered at risk for further kidney events after CPB due to preoperative eGFR values of ≥ 20 to < 60 mL/min/1.73 m2 and a minimum Society of Thoracic Surgeons (STS) Calculator Renal Failure Risk Score of ≥ 2.8%; the STS score will be calculated during the initial screening period. The STS Calculator is a model which utilizes patient characteristics and baseline clinical data to generate procedure-specific risk scores for patient outcomes [36, 37]. Participants are identified by qualified research staff via methods including review of medical records, external referrals, or use of databases. Recruitment strategies may include study posters, referral letters, recruitment brochures, advertisements, social media posts, and websites, where permitted by local regulations. All recruitment materials will be submitted to local institutional review boards/ethics committees for review and approval, as required, prior to use. This will be a global study conducted in at least 19 countries.
Following the initial 28-day screening period, randomization and dosing will occur within 7 days prior to surgery with CPB (Fig. 2). Participants will be assessed on Days 3, 7, 15, 30, 60, and 90 after surgery (primary evaluation period), with the primary endpoint analysis conducted at Day 90 post CPB (primary analysis). Serum creatinine (sCr) and serum cystatin C (sCysC) will be measured on the day of screening (baseline); day of dosing (day 1); day of surgery (prior to induction of anesthesia), daily from days 1 to 7; on days 15, 30, 60, and 90 post CPB; and at hospital discharge. Baseline eGFR is reported as the average eGFR calculated from the last assessment during the screening and day 1 visits, prior to study intervention administration. Participants will be contacted by telephone at one year post CPB to assess the requirement for KRT and survival (survival follow-up period). The total study duration is approximately 400 days. A participant will be considered to have completed the study once they have completed the primary evaluation period or withdrawn.
Fig. 2
ARTEMIS study design. *Weight-based dosing will ensure ravulizumab serum concentrations of ≥ 175 μg/mL and achieve complete terminal complement inhibition, while maintaining maximum ravulizumab serum concentrations below the highest observed value of 3000 μg/mL from all completed clinical studies, for ≥ 18 days in the target population
A global, multidisciplinary executive steering committee (ESC) of independent experts in the field was established in 2023. Since its inception, the ESC meets every 6 months, providing input into the ravulizumab CSA-AKI global clinical development program and acting in an advisory capacity to Alexion on the study. The study started in April 2023, and the current estimated completion date is February 2027.
Study endpointsThe primary endpoint in this study is the occurrence of MAKE90, defined as meeting at least one of the following events post CPB: initiation of KRT or death from any cause through day 90, or a decrease from baseline eGFR of ≥ 25% at day 90 calculated using the CKD-EPI equation and sCysC. This endpoint was chosen following discussions with and recommendations from both the Food and Drug Administration (FDA) and European Medicines Agency (EMA). The primary estimand is the population-level treatment difference between the two treatment groups in the proportion of participants experiencing MAKE90. The primary analysis will be based on the intent-to-treat (ITT) analysis set, which consists of all participants randomized to study intervention, under the treatment policy strategy. In this strategy, the values of the outcome of interest are used regardless of the occurrence of any intercurrent events. The treatment difference will be estimated using the Cochran-Mantel Haenszel (CMH) method adjusting for the two stratification variables (surgery type and baseline CKD stage). For the primary endpoint, missing data for KRT initiation or death will be imputed as a non-event, and missing eGFR values will be handled by multiple imputation in two steps. First, data will be imputed using Markov chain Monte Carlo to create a monotonic missing data pattern; then the second step will use the regression method for monotone missing data to impute the remaining missing observations. Rubin’s rule will be used for combining results to yield multiple imputation point estimates and standard errors. To minimize the risk of missing data, Alexion have a variety of patient retention initiatives including, but not limited to: home health and telehealth options for patients located substantial distances from clinical centers; clear and frequent communications provided to sites on the importance of complete data collection; active engagement with sites where patients are lost to follow-up to improve retention; and active site monitoring throughout the study. This allows for any issues to be addressed as they arise and prevents the development of more substantial complications.
Analysis of key secondary endpoints will also be based on the ITT analysis set and will include the occurrence and severity of Kidney Disease: Improving Global Outcomes (KDIGO)-defined AKI; length of post-operative intensive care unit stays; occurrence of KRT or mortality; and all-cause mortality. The primary assessment of MAKE90 will be based on sCysC measurements, as it is known that skeletal muscle atrophy (a common complication of cardiac surgery) can decrease sCr levels, leading to overestimation of eGFR after cardiac surgery [38, 39]. The secondary endpoints measuring AKI occurrence will be based on the sCr measurement. Additional secondary endpoints include MAKE assessed at days 30 and 60 post CPB using eGFR calculated from sCr or sCysC, MAKE assessed at day 90 post CPB using eGFR calculated from sCr, and occurrence of STS renal failure as determined by the Risk, Injury, Failure, Loss of kidney function, and End-stage kidney disease (RIFLE) Failure criteria. The proportion of participants meeting each of the key secondary endpoints will be summarized by treatment group. The point estimates and associated 2-sided 95% confidence intervals of the treatment difference in proportions will be estimated using the CMH method. Healthcare resource utilization will be assessed by documenting and comparing the length of total index hospital stays, number of days on mechanical ventilation, hospital readmissions, and days on KRT between treatment groups. Safety, pharmacokinetics and pharmacodynamics, and patient-reported outcomes of health-related quality of life will also be assessed. Exploratory endpoints in this study include exploration of the efficacy of ravulizumab in reducing the risk of CSA-AKI within 7 days of CPB, measured via sCysC (highest AKI stage by KDIGO criteria and/or occurrence of severe AKI [KDIGO stage 2 or 3]), and the assessment of biomarkers at baseline and following treatment. Biomarkers include, but are not limited to: complement pathway activation (plasma and urine sC5b-9), renal injury (urine neutrophil gelatinase-associated lipocalin), and endothelial damage (plasma thrombomodulin).
A series of subgroup analyses of the primary endpoint and key secondary endpoints are planned, including in the following participants: different races; different sexes; different age groups (18–60, 61–75, > 75 years); different CKD stages at baseline; and different surgical types during CPB.
Study assessments and proceduresStudy inclusion and exclusion criteria are listed in Table 1. The minimum Renal Failure Risk Score will additionally ensure that participants who are at risk for postoperative kidney events are enrolled [36, 37]. Baseline eGFR (and subsequent measures on days 30, 60, and 90) will be determined using the CKD Epidemiology Collaboration (CKD-EPI) equation based on central measurements of sCysC and/or sCr, as described above.
Table 1 Description of the key inclusion and exclusion criteria for enrollment in this studyApproximately 736 participants will be randomly assigned (1:1) using Interactive Response Technology (IRT) and the block randomization method to receive either ravulizumab or placebo, stratified by baseline CKD stage (3a, 3b, 4) and surgery type (mitral valve replacement or combined procedures vs other single procedure). The sample size estimation was based on a two-proportion difference using a normal approximation method to compare the treatment difference in the proportion of participants experiencing MAKE events within 90 days post CPB between the ravulizumab and the placebo group. A sample size of 736 (368 participants per treatment group) has at least 90% power to detect a statistically significant treatment difference of 10% in the proportion of participants with MAKE within 90 days post CPB under a 2-sided significance level of 0.05.
Participants, all investigative site personnel, and any Alexion employee or designee directly associated with the conduct of the study will be blinded to participant treatment assignment throughout the study. The blinding will be maintained by using identical study intervention kits, labels, and appearance of ravulizumab and placebo. The randomization code will be maintained by the IRT provider. In case of an emergency, the investigator has the sole responsibility for determining if unblinding of a participant’s intervention assignment is warranted, primarily based on participant safety. If the investigator decides that unblinding is warranted, the investigator may, at the investigator’s discretion, contact Alexion to discuss the situation prior to unblinding a participant’s intervention assignment unless this could delay emergency treatment for the participant. If a participant’s intervention assignment is unblinded, Alexion must be notified within 24 h after breaking the blind. The date and reason that the blind was broken must be recorded in the source documentation and case report form (CRF), as applicable.
In the case of unblinding, unblinded information will only be accessible to those who are involved in the safety reporting to Health Authorities, Independent Ethics Committees (IECs), Institutional Review Boards (IRBs), and/or the independent data monitoring committee (DMC). When unblinding is the result of an AE that is unexpected or related and serious, the blind will be broken for that specific participant only. In the case of unblinding (for example, due to safety concerns), persons responsible for ongoing study conduct and subsequent data analysis and interpretation will remain blinded.
Randomization should occur on the same day as dosing, except in cases when the study intervention needs to be prepared the day before dosing. Screening and randomization or randomization and surgery may occur on the same calendar day. Participants will receive a single weight-based intravenous dose of either placebo or ravulizumab on day 1 (Table 2) and surgery must be scheduled to occur within 7 days after dosing. In the event of unexpected surgical delay, surgery with CPB should occur no later than 15 days after study drug infusion. Study drug infusion must be completed at least 1 h prior to the surgery start. This schedule is based on predictions by model-based simulations that ravulizumab will provide complete inhibition of C5 for at least 18 days. These simulations combined data on serum concentrations of ravulizumab over time in patients with atypical hemolytic uremic syndrome [29] with data from two studies (NCT04543591 and NCT04557735) on the effect of blood transfusion on ravulizumab clearance in patients with hematopoietic stem cell transplantation–associated thrombotic microangiopathy.
Table 2 Weight-based single dose of ravulizumab or placebo given within 7 days prior to cardiac surgery with CPBEach 30-mL single-use vial of ravulizumab contains 300 mg of ravulizumab (10 mg/mL), 10 mM sodium phosphate, 150 mM sodium chloride, and 0.02% polysorbate 80 in water at pH 7.0. The placebo is formulated as a matching sterile solution with the same buffer components, without ravulizumab. All participants will receive investigator-determined standard of care in addition to the study interventions (according to institutional practices and participant characteristics) unless these are listed as prohibited concomitant therapies within the exclusion criteria (Table 1).
Data collection and managementAll participant study data will be recorded on the case report form (CRF) unless transmitted to Alexion or designee electronically. The investigator is responsible for verifying that data entries are accurate and correct by maintaining source data that supports the information in the CRF. The investigator must permit study-related monitoring, audits, IRBs/IECs review, and regulatory agency inspections, and provide direct access to source data documents. Alexion or designee is responsible for the data management of this study, including quality checking of the data. Records and documents, including signed informed consent forms, pertaining to the conduct of this study must be retained by the investigator for at least 25 years after study completion, or longer if required by local regulations or institutional policies. No records may be destroyed or transferred to another location/party without the written approval of Alexion. Clinical study documents and records required as part of the trial master file are archived and stored by Alexion for at least 30 years. Quality tolerance limits will be predefined to identify systematic issues that can impact participant safety and/or reliability of study results. These predefined parameters will be monitored during the study, and important deviations and remedial actions taken will be summarized in the clinical study report.
Monitoring of harmsAll adverse events (AE)s, including administration-related AEs, will be reported to the investigator or qualified designee by the participant (or surrogate when appropriate). The investigator is responsible for recording AEs or serious AEs (SAE) and remains responsible for following up on SAEs that are serious, considered related to the study, or cause the participant to discontinue. All AEs will be collected from the signing of the informed consent form until the end of the primary evaluation period; all SAEs will be recorded and reported to Alexion or the designee immediately, and under no circumstances should this exceed 24 h. Any updated SAE data will be submitted to Alexion or the designee within 24 h of becoming available.
Any dose of study intervention greater than that specified in the protocol will be considered an overdose. Accidental or suspected overdose without any association with laboratory abnormalities or clinical symptoms should not be considered an AE unless there are negative clinical consequences, but must be reported by the investigator to Alexion. No specific treatment for overdose is recommended.
Treatment-emergent AEs (TEAEs) are defined as AEs with onset (or existing events that worsen in severity) from the initiation of dosing of study intervention through 90 days post CPB. All AEs will be coded using the Medical Dictionary for Regulatory Activities, version 24.1 or higher, and will be summarized by System Organ Class and Preferred Term overall, by severity, and by relationship to study intervention. Detailed by-participant listings of all TEAEs (including those leading to withdrawal from the study) will be reported. Participants who experience a reaction during the administration of ravulizumab should be treated according to institutional guidelines. Alexion has insurance to cover the costs of research injuries as long as the participant followed the investigator’s instructions, the costs are reasonable, and the participant did not cause the injury.
Patients receiving complement inhibitors are at increased risk of infection by N. meningitidis. All participants in ARTEMIS must be vaccinated against N. meningitidis within the 3 years prior to enrollment or during the screening period; hospitalized participants may be vaccinated after study intervention administration but prior to hospital discharge. Vaccines against serotypes A, C, Y, and W135 (and serotype B where available if recommended by local guidelines) are recommended in preventing commonly pathogenic meningococcal serotypes. Participants must receive vaccination series as indicated according to current national vaccination guidelines. If ravulizumab dosing occurs less than 2 weeks after initial vaccination, participants will receive appropriate prophylactic antibiotics for at least 2 weeks after vaccination. All participants will also be monitored for early signs of meningococcal infection and treated if required. Furthermore, prior to study intervention administration, a safety card will be provided to participants to increase participant awareness of the risk of meningococcal infection, promote quick recognition of the signs/symptoms of infection, and inform participants on what actions must be taken if they are experiencing these.
Intravenous and infusion-related reactions are a potential risk with the use of monoclonal antibodies; these reactions can be nonimmune or immune mediated (e.g., hypersensitivity reactions). All administration, intravenous, and infusion-related reactions will be reported to the Investigator and qualified designee. The Investigator and qualified designee are responsible for detecting, documenting, and recording events that meet the definition of AE or SAE and remain responsible for following up on events that are serious, considered related to the study intervention, or study procedures; or that caused the participant to discontinue ravulizumab.
The primary analysis will occur when all enrolled participants have completed the primary evaluation period. An independent DMC, comprising experts in relevant fields with no direct relationship to the study, has been appointed by Alexion to assess safety and efficacy measures. The DMC will review unblinded study information provided by an independent data analysis center during the conduct of the study for periodic safety review (at least quarterly) and two planned interim analyses. The two interim analyses are planned after approximately 30% and 50% of the randomized participants have completed the primary evaluation period (i.e., day 90 post CPB Visit). At each interim analysis, the DMC will make formal recommendations based on the pre-specified criterion contained in the DMC statistical analysis plan. The purpose of the first interim analysis is to assess early futility, while the second interim analysis will include futility, early efficacy, and sample size re-estimation assessments. Any amendments to the protocol will be submitted for IRB/IEC and health authority approval before the implementation of changes made to the study design, except for changes necessary to eliminate an immediate hazard to study participants. All amendments to date will be reported in the amended protocol document, which will be provided to all study sites/investigators as well as updated on all relevant clinical trial registries. Following any protocol amendments, specific training will be conducted with sites to ensure compliance. When a protocol deviation is identified—for example, during the active monitoring of sites conducted by Alexion and its appointees—a thorough deviation standard operating procedure (SOP) is in place to investigate, classify, report, and mitigate the identified issue(s).
Comments (0)