Fatal Late-Onset Presentation of Ornithine Transcarbamylase (OTC) Deficiency: A Case Report Highlighting Diagnostic Challenges and Cerebral Complications
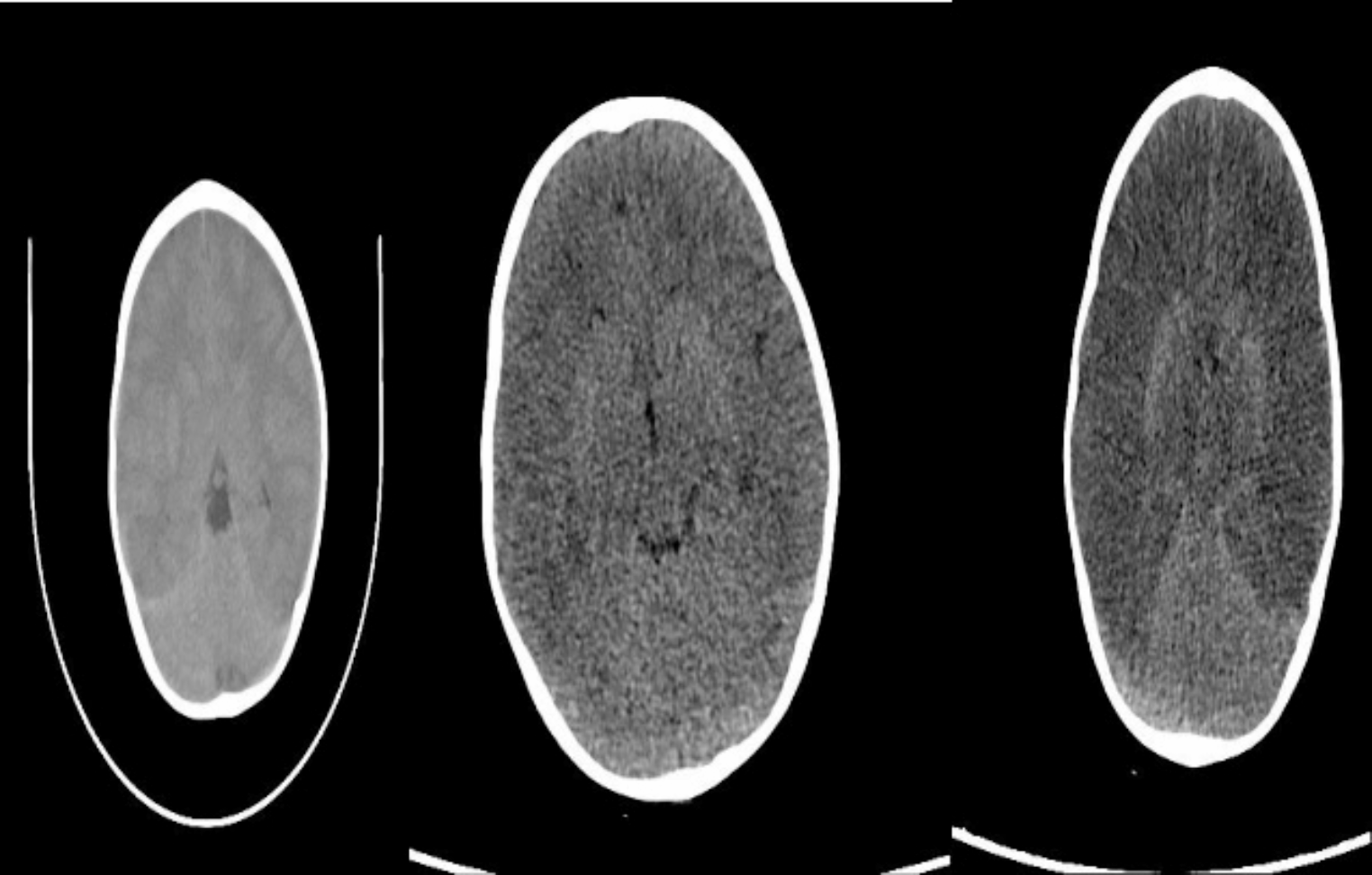
Ornithine transcarbamylase deficiency caused by mutations in the OTC gene is the most common inherited disease of the urea cycle, and it has a variable phenotype. This inherited disorder is associated with hyperammonemia. OTC deficiency; usually occur in the neonatal period and its incidence is relatively rare in childhood. Here, we present a case to illustrate the late-onset manifestations of OTC deficiency, underscoring its potentially fatal consequences if not promptly recognized and the critical importance of early diagnosis. A 2 years old male, presented with three days of progressive symptoms, including daily weakness, loss of appetite, and vomiting. Following a deterioration in his level of consciousness, he was transferred to our hospital after receiving intravenous hydration at another facility. Upon admission, the patient exhibited vomiting, impaired consciousness, hyperammonemia, and mild metabolic acidosis. Imaging studies, including computed tomography and magnetic resonance imaging, revealed cerebral edema but no other significant abnormality. His blood ammonia level was alarmingly high at 987 µg/dl. The patient was immediately admitted to the intensive care unit, intubated, and treated for cerebral edema with continuous hemodiafiltration. Although his ammonia levels decreased after 24 h of treatment, his cerebral edema worsened significantly. Despite aggressive management, including the administration of Carbaglu, L-arginine, and prolonged hemodialysis, the patient succumbed to cerebral death due to worsening cerebral edema. Genetic analysis revealed a c.586G > A (p.Asp196Asn) mutation. High clinical suspicion for a urea cycle disorder is essential for timely diagnosis and appropriate treatment in patients of any age presenting with encephalopathy and persistent vomiting.

Comments (0)