Clinical, Biochemical and Molecular Findings of Two PMM2-CDG Cases from India
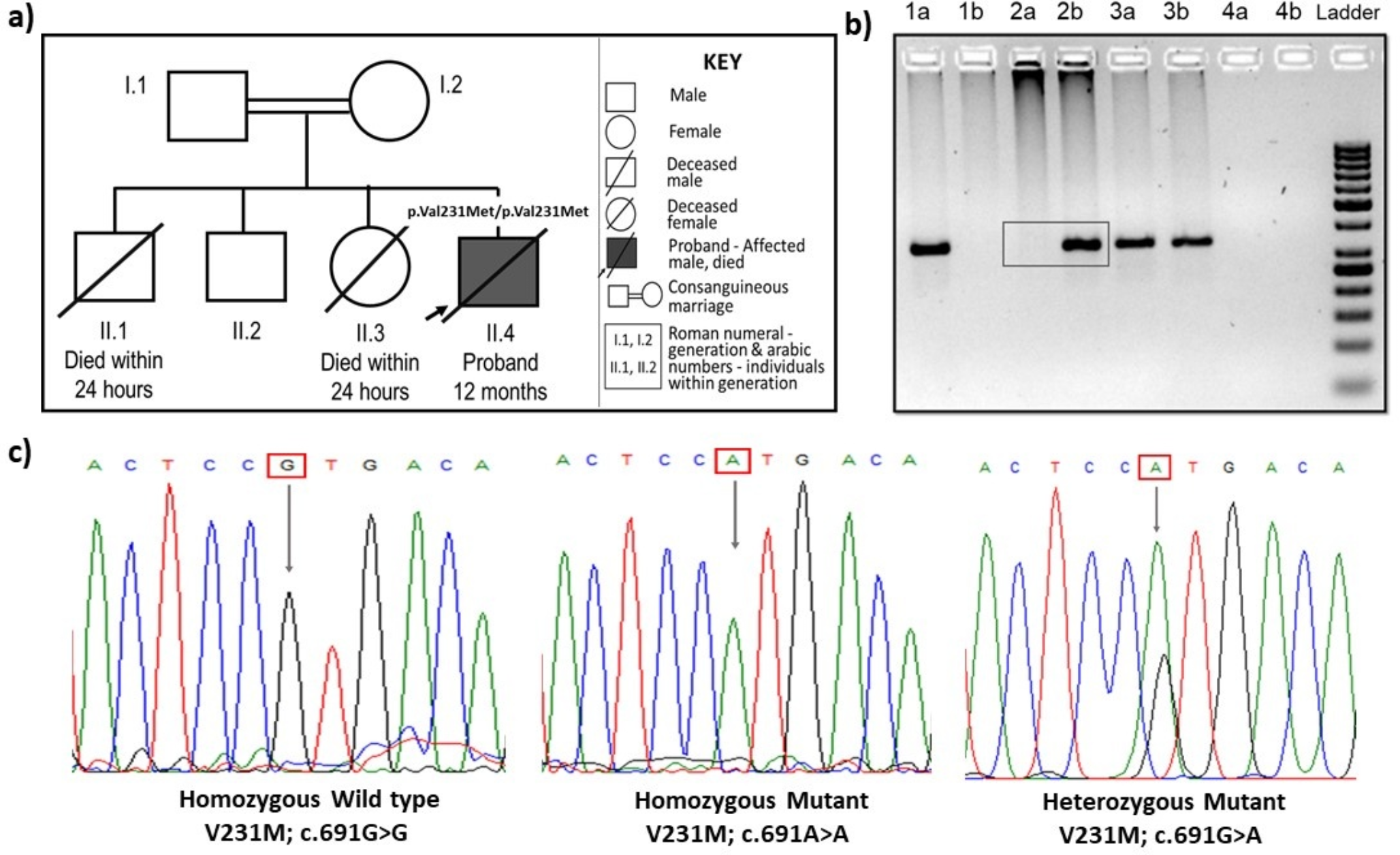
Congenital Disorders of Glycosylation (CDG) represent a diverse and complex group of inherited metabolic disorders, with more than 170 distinct types identified to date. Among these, the most common and first characterized is PMM2-CDG, which results from mutations in the PMM2 gene responsible for encoding phosphomannomutase. This disorder follows an autosomal recessive inheritance pattern. Since the initial identification of PMM2-CDG in 1980, over 1,000 patients have been documented globally. Diagnosis usually involves transferrin isoform analysis, which typically reveals a type I pattern, alongside molecular confirmation of biallelic pathogenic variants that are distributed throughout the PMM2 gene. Till date, only two patients of PMM2-CDG have been reported from India, and both these patients exhibited unusual presentations. However, recent economic feasibility of genome sequencing along with the availability of transferrin isoform analysis in India, have made it possible to identify this often underdiagnosed disorder more effectively. Here, we report two molecularly confirmed PMM2-CDG patients who presented with classic phenotypes, biochemical evaluations and notable family histories. Given the severity of the disease and the limited treatment options currently available, early diagnosis of affected children and the identification of mutations for prenatal testing are critical. This approach not only helps to alleviate the disease burden but also enables timely interventions within our population, ultimately improving patient outcomes.

Comments (0)