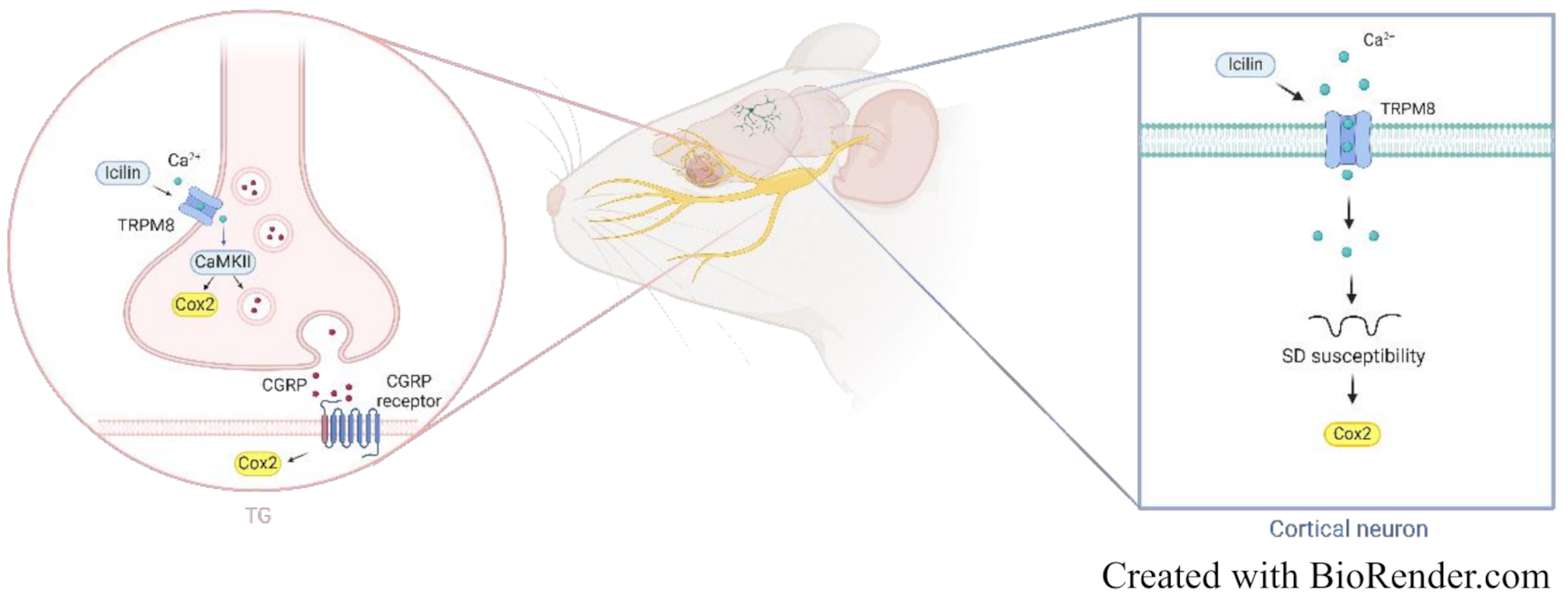
This study provides novel insights into the potential role of TRPM8 in migraine the by exploring its central effects on SD and its peripheral effects on TG neuronal activity and inflammation. By demonstrating that activation of cortical neuronal TRPM8 increases susceptibility to SD and SD-evoked cortical neuroinflammation, possibly independent of intrinsic regulatory mechanisms, our findings shed light on previously unexplored central aspects of cortical TRPM8 function. Furthermore, our results also address the peripheral contribution, specifically, trigeminal TRPM8 activation, to the release and expression of CGRP and trigeminal neuroinflammatory responses via a CaMKII-dependent mechanism. Collectively, these findings underscore the the central and peripheral TRPM8 functions, providing insights into possible therapeutic strategies targeting TRPM8 for migraine.
Potential mechanisms underlying TRPM8’s influence on SD, TG neuron, and migraine-related behaviors
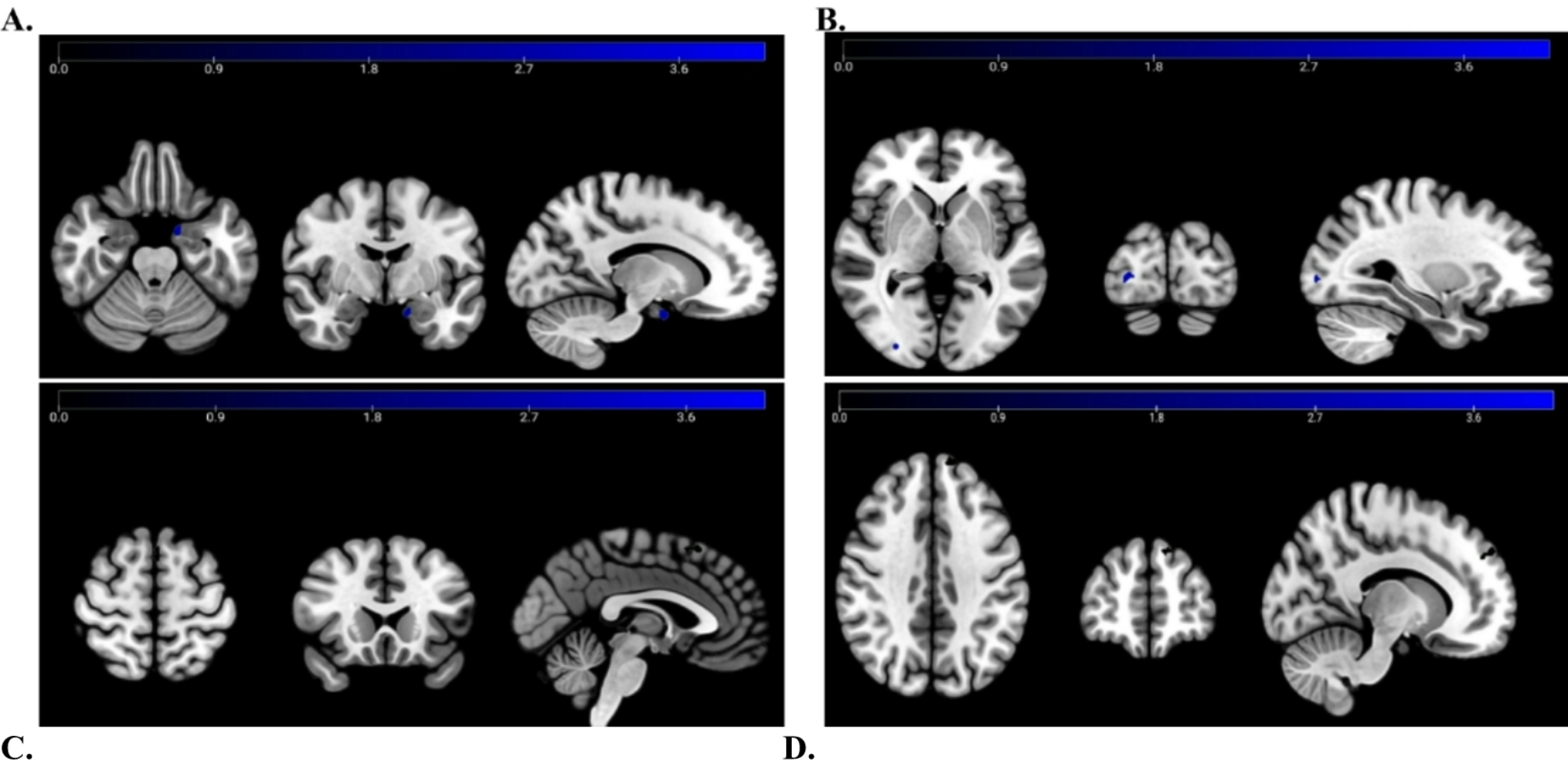
To the best of our knowledge, this study is among the first to explore the potential relationship between TRPM8 and cortical SD (Fig. 2B and C). Our findings suggest that TRPM8 activation may increase the frequency of SD events. Elevations in extracellular glutamate and K+ are crucial for initiating and propagation of SD. It is plausible that TRPM8 activation influences SD susceptibility by increasing intracellular Ca2+ levels, which could contribute to neurotransmitter release. Studies indicate that menthol-induced TRPM8 activation leads to glutamate release from DRG neuronal terminals [42,43,44]. Furthermore, TRPM8 has been reported to activate large-conductance Ca2+-activated K+ channels (BK channels), promoting the outward K+ flow upon Ca2+ influx [45]. Notably, TRPM8 activation has been linked to increased neuronal excitability by inhibiting leak K+ conductance [46], potentially accounting for the depolarized resting membrane potential observed after SD. These findings suggest that TRPM8 activity may modulate susceptibility to SD by affecting excitatory neurotransmission, K+ efflux, and intrinsic neuronal membrane properties.
The propagation of SD is critically dependent on Ca2+ influx. Previous studies have shown that SD triggered by localized high K+ or brief electrical stimulation requires Ca2+ for its propagation [47, 48]. For example, in rat neocortical slices, pre-treatment with Ca2+-free media completely prevented SD induced by localized high K+ exposure [49]. Similarly, non-selective Ca2+ channel blockers have been shown to prevent SD propagation in hippocampal slices following high K+ exposure, further supporting the notion that calcium influx is essential for SD spread [50]. Studies in genetically modified mice with mutations in the P/Q-type Ca2+ channel gene (CACN1A) provide additional evidence for the crucial role of calcium in SD propagation [51,52,53]. Gain-of-function knock-in mutations of the Cacna1a gene have been associated with reduced SD thresholds and accelerated propagation speed, underscoring the potential involvement of P/Q-type channels in SD regulation. Since P/Q-type Ca2+ channels mediate calcium-dependent neurotransmitter release [52], both non-selective Ca2+ channel blockers and selective inhibitors of P/Q-type channels effectively block SD propagation [53], further emphasizing the essential role of Ca2+influx in SD propagation.
CaMKII, a serine/threonine kinase, is activated in response to elevated intracellular Ca²⁺ levels [54]. In our TG primary culture experiments, we observed that TRPM8 activation leads to CaMKII activation (Fig. 3C). While this mechanism has not been directly examined in the cerebral cortex, it is reasonable to hypothesize that TRPM8 in the cortex may operate through pathways similar to those identified in the TG. Although direct evidence linking CaMKII to SD susceptibility is lacking, previous studies have reported that SD triggers transient and reversible widespread fission of the cortical endoplasmic reticulum (ER). Furthermore, CaMKII inhibition has been shown to mitigate SD-induced neuronal activity suppression and prevent ER fission [55]. TRPM8 activation may elevate intracellular Ca²⁺ levels, which may potentially initiate downstream signaling events such as CaMKII activation. We speculate that this cascade could play a role in SD-related events, such as CaMKII-mediated ER fission and associated neuronal dysfunction.
Previous studies have explored the potential role of TRPM8 in pain or migraine-related behaviors, including the mechanical allodynia in nitroglycerin (NTG)-induced chronic migraine model, but the results have been inconsistent, likely influenced by various factors. The function of TRPM8 appears to depend on the context of injury. Under cold stimuli, TRPM8 mediates cold allodynia by regulating neuronal activity [43, 45, 46]. However, when exposed to stronger noxious stimuli, such as formalin or inflammatory soup, TRPM8 exhibits anti-nociceptive effects [23, 44]. In the NTG-induced chronic migraine model, conflicting results have also been observed. TRPM8 lowers the mechanical allodynia threshold during the NTG administration phase [18] but facilitates the recovery of allodynia thresholds following repeated NTG injections [24]. These findings suggest that TRPM8 may contribute ro mechanical hypersensitivity in the presence of noxious stimuli. However, in the absence of noxious stimuli, TRPM8 plays a protective role in promoting recovery from hypersensitivity through endogenous testosterone, an endogenous ligand of TRPM8 [24]. Taken together with the effects observed in other animal models, we speculate that the role of TRPM8 in migraine-related mechanisms is not limited to a single pathway but likely involves contributions at multiple mechanisms, including modulating SD and nociceptive mechanisms.
TVS activation is pivotal in migraine pathogenesis. While the precise mechanism underlying TVS activation remains incompletely elucidated, meningeal vasodilatation response, meningeal inflammation, and sensitization of TNC neurons are critical components of TVS activation. A study using intravital microscopy showed that TRPM8 activation by menthol altered the basal meningeal arterial tone, causing meningeal vasodilation [56]. In an ex vivo hemiskull model, menthol stimulated CGRP release from the hemiskull, TG, and TNC [57]. Furthermore, electrophysiological recordings demonstrate that menthol increased both excitatory and inhibitory synaptic transmission in the TCC [56, 58]. These findings raise a possibility that potential pronociceptive role of TRPM8.
Characterization of TRPM8 expression and activity
In our experiments, to minimize non-specific TRPM8 signals, we utilized a TRPM8 primary antibody that has been validated for specificity in knockout cells [63]. Our results show that the TRPM8 signal in TG cultures appeared relatively faint compared to markers such as beta III tubulin (Additional file 3D), likely reflecting the low endogenous expression of TRPM8 in these neurons under our experimental conditions. Our quantitative results revealed that approximately 50% of the culture consisted of neurons (Additional file 3 C), with around half of these neurons expressing TRPM8 (Additional file 3D), suggesting that TRPM8-positive cells accounted for roughly 25% of the total TG cell population. In contrast, previous studies using immunohistochemistry and in situ hybridization in rats and mice reported that TRPM8 is expressed in 11.8% of adult TG neurons [59] and 5–10% in adult DRG neurons [60], slightly lower than our finding (Additional file 3 C-D). The observed difference may be attributed to variations in experimental methodologies—ours is based on primary cultures, whereas previous studies typically employed tissue immunohistochemistry. Additionally, developmental dynamics of TRPM8 expression likely contribute to this discrepancy. TRPM8 expression follows a biphasic pattern during development [61]. It begins at E14 and increases significantly from E14 to E18 [61, 62], peaks at birth, and then declines by P4. A second wave of upregulation occurs between P4 and P12, followed by a slight reduction and eventual stabilization in adulthood [61]. The higher percentage of TRPM8-positive neurons observed in our primary cultures—around 25%, compared to approximately 5–11% in the literature—may stem from the fact that our cultures were derived from P8-P10 rat pups, which corresponds to the second developmental wave of TRPM8 upregulation. In contrast, several studies use adult rats, where TRPM8 expression in both the TG (the location of cell bodies of primary afferent neurons) and the afferent fibers projecting to the meninges typically shows a decline compared to earlier developmental stages [9, 61].
The subcellular localization of TRPM8 was clarified by examining its distribution in relation to the cytoskeletal marker beta III tubulin. In the overlay images, circular hollow regions devoid of beta III tubulin staining correspond to the nuclei of the cells(Additional file 3D), as confirmed in a recent published study regarding TG primary culture [63]. The absence of TRPM8 signal in these nuclear regions indicates that TRPM8 does not localize to the nucleus, consistent with findings from recent studies [59]. A comparison of the single-channel TRPM8 staining image with the overlay image of beta III tubulin further supports the conclusion that TRPM8 is localized in the cytoplasm rather than the nuclear region.
Additionally, our TRPM8 staining in TG primary cultures exhibited a relatively uniform expression pattern, which may be attributed to the controlled cell seeding protocol used. Uneven seeding can result in variability in cell behavior, including differences in cell proliferation, differentiation, and cell-cell communication [64,65,66]. Maintaining consistent seeding density is crucial for obtaining reliable results, particularly in biomolecular assays, such as those analyzing supernatants and components isolated from cell lysates [67, 68]. However, while our primary cultures exhibit a relatively uniform TRPM8 expression pattern, this may not fully reflect the in vivo distribution of TRPM8 in TG tissue. In tissue sections, TG cells typically exhibit an uneven distribution, as demonstrated in previous studies [33, 36, 59]. The uniform distribution of TRPM8 in our primary cultures may be a consequence of the controlled seeding process, which contrasts with the more heterogeneous expression pattern seen in tissue. Therefore, the TRPM8 distribution in our cultured TG neurons may not directly mirror the expression pattern in intact TG tissue. Regarding TRPM8 expression, most studies have focused on its presence in the peripheral nervous system, including TG and DRG [59]. To the best of our knowledge, TRPM8 expression in the brain has primarily been documented using techniques such as western blot [10] or in situ hybridization [11]. Here, we provide the first evidence of TRPM8 protein expression in the cerebral cortex, highlighting its spatial distribution. TRPM8 is extensively expressed in cortical neurons, accounting for 66.2–84.5% of the neuronal population (Fig. 1 & Additional file 1), which is notably higher than the expression levels observed in TG primary neurons. TRPM8 expression in both the cortex and TG is primarily localized to neurons. Despite this similarity, the distribution of TRPM8 differs between the two tissues. In the cortex, TRPM8 is more uniformly expressed across cortical neurons, whereas in intact TG tissue, it shows a more uneven distribution. This difference likely reflects the distinct spatial distribution pattern and neuronal organization in each tissue.
TRPM8 is a Ca²⁺-permeable channel. In our study, we measured intracellular Ca²⁺ levels using the 340/380 nm excitation ratio for Fura-2 as an indicator of TRPM8 activation. Our results showed a modest increase in the ratio from 0.30 to 0.35, indicating a small change of 0.05 following icilin treatment. In contrast, a previous study using stable TRPM8-expressing cell lines reported a much larger Δ340/380 change of approximately 3 to 4 at the same icilin concentration (10 μM) [69]. A significant challenge in our study was the limited cell yield from TG primary cultures, which required pooling cells for functional assays. Additionally, as noted earlier, the endogenous expression of TRPM8 in both TG primary cultures and intact TG tissue is low, which made Ca²⁺ level measurements particularly challenging. The TRPM8-expressing cell lines used in previous studies showed high TRPM8 expression (though the exact percentage was unspecified, images indicated a high proportion of transfected cells) [69], and our findings likely reflect the lower TRPM8 expression levels inherent to our experimental conditions. This difference may account for the relatively small response observed in our study.
Our findings show that after icilin treatment, the Ca²⁺ concentration rises and remains elevated above baseline for at least 500 s (Fig. 3B). However, these changes may not fully reflect calcium channel activity. A previous study demonstrated that while Ca²⁺ current changes return to baseline within approximately 5 s under continuous icilin exposure, intracellular Ca²⁺ concentrations remain elevated for up to 100 s [69]. Although the previous study did not assess longer-term changes in intracellular Ca²⁺ levels (i.e., beyond 100 s), their results, showing no noticeable recovery in Ca²⁺ concentration, are consistent with our findings (Fig. 3B). This suggests that intracellular Ca²⁺ dynamics may not entirely capture the transient nature of Ca²⁺ currents. Ca²⁺ currents, driven by the rapid movement of ions across the membrane, are tightly regulated by membrane potential and typically last milliseconds to seconds. In contrast, intracellular Ca²⁺ dynamics involve more complex processes, such as Ca²⁺ entry, release from intracellular stores, buffering by cytosolic proteins, and diffusion, which sustain changes for tens to hundreds of seconds [70], which contribute to sustained changes lasting tens to hundreds of seconds. Importantly, these sustained changes do not necessarily diminish the functional relevance of the initial Ca²⁺ rise above baseline observed after icilin treatment (Fig. 3B). On the contrary, they highlight the complexity of evaluating Ca²⁺ channel activity based solely on intracellular Ca²⁺ levels. The duration of the elevated Ca²⁺ state may be influenced by buffering mechanisms or the diffusion rate of Ca²⁺ indicators, which could limit its ability to fully reflect ion channel activity compared to current recordings. Therefore, the prolonged intracellular Ca²⁺ responses are likely driven by downstream processes, rather than continuous TRPM8 activity, indicating a limitation of the calcium imaging technique. Nonetheless, these prolonged responses do not negate the physiological significance of the initial Ca²⁺ rise induced by icilin.
Possible mechanisms regulating TRPM8 expression and activity
We present novel evidence demonstrating that TRPM8 exhibits a nearly homogeneous distribution in the cerebral cortex (Additional file 1) and demonstrate that SD downregulates TRPM8 expression (Additional file 2B). TRPM8 expression or activity is modulated by several molecules, including PIP2 [71] and protein kinase C (PKC) [72,73,74]. SD has been reported to enhance PKC activity in the cerebral cortex [75] and PKC-mediated desensitization of TRPM8 has been documented [72,73,74]. Therefore, SD-induced PKC activation may plausibly contribute to the observed reduction in TRPM8 expression following SD. Additionally, F11, a TRPM8 agonist, has been shown to increase TRPM8 expression on the plasma membrane [76], which could potentially rescue TRPM8 expression following SD induction, and might explain why TRPM8 expression remained unchanged when icilin treatment was followed by SD induction (Additional file 2D). The lack of significant changes in TRPM8 expression after icilin treatment alone (Additional file 2 C) may be attributed to this transient, agonist-dependent recruitment of TRPM8. From a pathophysiological perspective, our findings suggest that while TRPM8 may facilitate susceptibility to SD, the SD-induced downregulation of TRPM8 could serve as a compensatory mechanism to mitigate the effects of TRPM8 activation. Currently, there is a lack of literature explaining why TRPM8 expression is downregulated after SD induction. However, based on our findings suggesting a potential compensatory mechanism, future experiments could aim to investigate this further. One approach could involve utilizing optogenetic techniques to induce repeated SD events in awake animals. This method would allow the assessment of SD susceptibility at multiple time points after the initial induction, including 2 h or longer. By performing repeated SD inductions in awake animals, it could be determined whether the observed downregulation of TRPM8 leads to reduced susceptibility to SD in subsequent tests, which would help clarify the role of TRPM8 in modulating SD.
GWAS have identified a significant association between specific SNPs within the TRPM8 gene, specifically rs10166942[C/T] and rs17862920[T/C], and an increased susceptibility to migraine. Importantly, analysis of TRPM8 mRNA expression in the DRG has revealed lower expression level associated with the chromosome harboring rs10166942[C], in contrast to the chromosome carrying the rs10166942[T] variant [6]. Individuals bearing C-allele in the rs10166942 variant exhibit a significantly reduced sensitivity to cold pain compared to non C-allele carriers [6], while those the rs10166942[T] allele exhibit an increased risk of migraine [3,4,5]. Moreover, migraine patients with the rs10166942[T] allele are predisposed to chronic migraine and allodynic symptoms [5]. This observed association suggests that genetic polymorphisms could influence TRPM8 expression and activity, and potentially contributing to an increased susceptibility to migraine.
TRPM8 and neuroinflammation
Animal studies suggest that SD triggers the opening of pannexin 1 (PANX1), facilitating the formation of the P2 × 7–PANX1 pore complex. This complex enables subsequent activation of the P2 × 7 receptor, which plays a crucial role in SD-induced neuroinflammation [36, 39, 77, 78]. Although there is no direct evidence explaining how TRPM8 activation by icilin treatment enhances SD-induced cortical neuron inflammation, purinergic P2 × 7-mediated mechanisms may still be involved. TRPM8 activators have been shown to increase ATP release [79]. Additionally, inhibition of pannexin 1 suppresses TRPM8-induced ciliary beat frequency, suggesting that purinergic receptors might be part of the downstream signaling pathway activated by TRPM8 and could contribute to its effects.
Regarding the relationship between SD and TG inflammation, we previously examined Cox2 expression in the TG following SD, but found no significant change in Cox2 expression after CSD [







Comments (0)