Diabetes is mainly composed of type 2 diabetes (T2D) (90%), pathologically characterized by insulin resistance and beta cell dysfunction, which leads to insulin release and abnormal blood glucose levels, triggering inflammation and endothelial dysfunction, and damaging the vasculature of the kidneys, eyes, heart, and nerves.1 2 Currently, oral hypoglycemic agents are the main treatment for T2D, but the side effects resulting from frequent administration have greatly undermined the treatment efficacy.3 Glycolipids are the main sources of energy in the human body, and the disorders in glycolipid metabolism are closely associated with the pathogenesis of T2D.4 Common symptoms of disordered glycolipid metabolism include decreased glucose uptake and insulin utilization efficiency, leading to insulin resistance and oxidative stress, which further triggers chronic inflammation in organs.5 Notably, molecular targeted therapy has been widely cited and shown significant therapeutic effects in the management of disordered glycolipid metabolism.6 7 This study explored the mechanism of glycolipid metabolism in T2D and aimed to identify potential therapeutic approaches for T2D by alleviating glycolipid metabolism disorders.
TGFB-induced factor homeobox 1 (TGIF1) is a multifunctional protein involved in gene transcription and activation.8 TGIF1 is an important contributor to diabetes and its complications. For instance, TGIF1 inhibits transforming growth factor beta (TGF-β) signaling and alleviates cardiac fibrosis in mice with diabetes.9 In diabetic nephropathy, upregulation of the transcriptional activity of TGIF1 can suppress kidney inflammation and fibrosis.10 Of note, increasing evidence has suggested that stable expression of TGIF1 is closely associated with adipogenesis and insulin function.11 12 Another report has found that TGIF1 is potentially related to the inhibition of lipid synthesis in lipid metabolism.13 Currently, no research has revealed the mechanism of TGIF1 in hepatic glycolipid metabolism in the pathogenesis of T2D.
MicroRNAs (miRNAs/miRs) participate in post-transcriptional regulation of gene expression and serve as critical regulatory factors in cell growth, differentiation, development, and apoptosis.14 Given the high stability and reproducibility of miRNAs in tissues and body fluids, their function as biomarkers for T2D is considered feasible and valuable.15 Importantly, recent studies have found that miRNAs are involved in the pathogenesis of T2D, including impaired insulin secretion and abnormal glycolipid metabolism.5 In particular, an analysis has shown that miR-106-5 p is poorly expressed in T2D.16 Our study further analyzed the upstream and downstream mechanisms of miR-106-5 p in T2D.
The early growth response (EGR) transcription factors play a significant role in controlling insulin biosynthesis, glucose homeostasis, inflammation, and pancreatic β-cell size.17 Early growth response 2 (EGR2), as a member of the EGR proteins, plays a positive regulatory role in adipocyte differentiation and inflammation, which potentially links to insulin resistance.18 Moreover, EGR2 expression is upregulated in a mouse model of T2D and exacerbates insulin resistance in hepatocytes.19 While silencing EGR2 expression can accelerate hepatic glucose uptake and restore normal lipid metabolism.20 Accordingly, we hypothesized that targeting EGR2 may have therapeutic potential for alleviating the dysregulation of glycolipid metabolism in T2D.
In this study, we investigated the regulatory mechanism of TGIF in the glycolipid metabolism of mice with T2D through the miR-106b-5p/EGR2 axis, aiming to identify new targets for the treatment of diabetes and provide a theoretical basis.
Materials and methods
Laboratory animals
The male C57BL/6J mice purchased from Phenotek (Shanghai, China, license number: SYXK (Hu) 2023-0038) were housed under standard conditions of constant temperature (22±3℃), 60% humidity, and a 12-hour light/dark cycle, with ad libitum access to food and water.
T2D model establishment and treatment
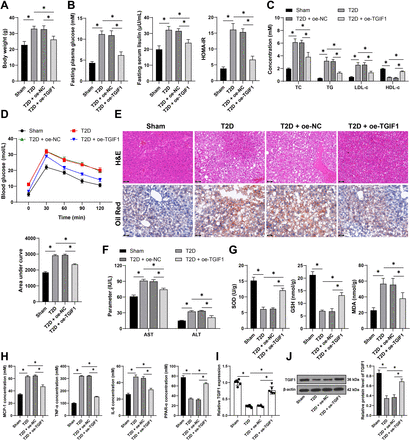
A total of 72 C57BL/6J mice (age 6–8 weeks) were randomly allocated into the following groups using the random number table method: sham group, T2D group, T2D+oe NC group, T2D+oe-TGIF1 group, T2D+oe-TGIF1+oe NC group, and T2D+oe-TGIF1+oe-EGR2 group, with 12 mice in each group. The corresponding lentiviral vectors (2×107 TU) were injected into each group of mice for 3 consecutive days. The oe-NC, oe-TGIF1, and oe-EGR2 lentiviruses were provided by GeneChem (Shanghai, China). After the injections, the mice in the T2D group were fed a high-fat diet (XTHF60, Xietong Pharmaceutical Bio-engineering, Jiangsu, China) for 6 weeks and then received intraperitoneal injections of streptozotocin (STZ) (45 mg/kg, S0130, Sigma, St. Louis, Missouri, USA) for 3 consecutive days to establish the T2D mouse model, with fasting plasma glucose (FPG) >11.1 mM indicating successful model establishment. The mice in the sham group were fed a normal diet (XT93M, Xietong Pharmaceutical Bio-engineering) for 6 weeks and received intraperitoneal injections of citrate buffer (3 mL/kg) for 3 consecutive days. After the feeding, related indicators were measured, and the mice were euthanized using 2% isoflurane anesthesia.21 In each group, liver tissues were collected from six randomly selected mice for histological experiments, and another six mice were used for molecular experiments.
Isolation, treatment, and modeling of primary mouse hepatocytes
The mouse liver was perfused with Hanks solution containing collagenase. After removal from the abdominal cavity, the liver was transferred to a sterile 6 cm culture dish and minced with sterile forceps. The suspension was then filtered through a 70 µm sieve, and the hepatocytes were separated by Percoll density centrifugation. Primary mouse hepatocytes were cultured in Dulbecco’s modified Eagle medium (Gibco, Carlsbad, California, USA) containing 10% fetal bovine serum (10099158, Gibco) and 1% penicillin/streptomycin (15140148, Gibco). The cells were incubated with 0.1 mM palmitic acid (PA) (P0500, Sigma)22 for 24 hours to establish a model of glycolipid metabolism disorder. The control group was normal hepatocytes.
Cell transfection
The miR-106b-5p mimic, mimic NC, miR-106b-5p inhibitor, inhibitor NC, TGIF1 overexpression plasmid (oe-TGIF1), EGR2 overexpression plasmid (oe-EGR2), and negative control plasmid (oe-NC) were provided by GeneChem. Cells were transfected with RNA or plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, California, USA), and the transfection efficiency was determined 48 hours post-transfection. Subsequently, the cells were induced to establish a model of glycolipid metabolism disorder.
Measurement of FPG, insulin, and Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) index
After 4 weeks of feeding, mice were fasted overnight for 12 hours, and blood samples were collected from the tail vein to measure FPG and fasting insulin (FINS) levels. Blood glucose levels were determined using an Accu-check glucometer (Roche, Basle, Switzerland). Insulin levels were measured using a mouse insulin ELISA kit (ab277390, Abcam, Cambridge, Massachusetts, USA) according to the manufacturer’s instructions. The HOMA-IR index was calculated using the formula: HOMA-IR = (FPG mmol/L×FINS μU/mL)/22.5.
Serum biochemical analysis
The levels of blood lipids, including total cholesterol (TC), triglycerides (TG), low-density lipoprotein cholesterol (LDL-c), and high-density lipoprotein cholesterol (HDL-c), were measured using a fully automatic biochemical analyzer (Chemray 240, Rayto, Shenzhen, China).
The levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in the serum were determined using a mouse AST ELISA kit (ab263882, Abcam) and a mouse ALT ELISA kit (ab282882, Abcam) according to the manufacturer’s instructions.
The levels of superoxide dismutase (SOD), glutathione (GSH), and malondialdehyde (MDA) in the serum were measured using a mouse SOD ELISA kit (ml643059, mlbio, Shanghai, China), a mouse GSH ELISA kit (ml063305, mlbio), and a mouse MDA ELISA kit (E-EL-0060, Elabscience, Shanghai, China) following the manufacturer’s instructions.
Glucose tolerance test
The glucose tolerance test was performed 4 weeks after the establishment of the T2D model. Mice fasted overnight for 12 hours and then were intraperitoneally injected with glucose (2 g/kg). Blood samples were collected from the tail vein at 0, 30, 60, 90, and 120 min after the glucose injection to measure blood glucose levels. GraphPad Prism V.8.0 software (GraphPad Software, San Diego, California, USA) was used for plotting and calculating the area under the curve (AUC), where a larger AUC value indicates poorer glucose tolerance.
Histopathologic analysis
The liver tissues of mice were fixed in formalin and embedded in paraffin. Sections of 4 µm thickness were prepared and stained with H&E staining for histologic examination. For the analysis of hepatic lipid accumulation, liver tissues were embedded in frozen sections, and 10 µm thick sections were stained with Oil Red O.
Measurement of cytokine levels
Mouse monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6) levels were determined using the mouse MCP-1 ELISA kit (ab208979, Abcam), mouse TNF-α ELISA kit (ab208348, Abcam), and mouse IL-6 ELISA kit (ab222503, Abcam) according to the manufacturer’s instructions. Additionally, mouse peroxisome proliferator-activated receptor alpha (PPAR-α) levels were measured using the mouse PPAR-α ELISA kit (ml002182, mlbio).
Quantitative real-time PCR (qRT-PCR)
Extraction of total RNA was performed using TRIzol Reagent (15596018CN, Invitrogen). The extracted RNA was synthesized to first-strand complementary DNA (cDNA) employing the PrimeScript RT Reagent Kit (RR047A, Takara, Dalian, China). For miRNA quantification, cDNA was synthesized using the Mir-X miRNA first-strand synthesis kit (638315, Takara). qRT-PCR was performed using Premix Ex Taq (RR390A, Takara), with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or U6 as the endogenous control.23 The relative expression levels of RNA were calculated using the 2-ΔΔCt method.24 Primer sequences are listed in table 1.
Table 1
View inline•Open as popup
qPCR primers
Western blot assay
Proteins were extracted from liver tissue and cells using a protein extraction kit (BC3710, Solarbio, Beijing, China). Proteins were then separated on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane. The membrane was blocked with 5% non-fat milk and incubated overnight at 4°C with primary antibodies of rabbit anti-TGIF1 (1:1000, PA5-102880, Thermo Fisher Scientific, Waltham, Massachusetts, USA), rabbit anti-EGR2 (1:1000, PA5-27814, Thermo Fisher Scientific), and rabbit anti-β-actin (1:1000, PA1-183-HRP, Thermo Fisher Scientific). After washing, goat anti-rabbit immunoglobulin G (IgG) secondary antibody (1:10,000, 31460, Thermo Fisher Scientific) was added. Target protein expressions were detected with ECL Western blotting substrate (PE0010, Thermo Fisher Scientific) and β-actin served as an internal reference.
Cell counting kit-8 (CCK-8) assay
Cells were seeded at 3×103 per well in a 96-well plate. After 48 hours, 10 µL of CCK-8 reagent (CK04, Dojindo Laboratories, Tokyo, Japan) was added to each well and the plate was incubated for 3 hours. Absorbance was then measured at 450 nm using a microplate reader (Bio-Rad, Hercules, California, USA).
Glucose uptake, consumption, and glycogen content measurement
Cells were seeded at 1500 cells per well in 96-well plates. Glucose uptake by hepatocytes was measured using the glucose uptake assay kit (ab136955, Abcam) following the manufacturer’s instructions.
A total of 1×105 cells were seeded in 24-well plates. On reaching confluence, the medium was switched to a starvation medium containing 10 mM glucose. After culturing for 48 hours, the glucose concentration in the medium was measured using the glucose assay kit (ab65333, Abcam) according to the manufacturer’s guidelines. Glucose consumption was calculated by measuring the glucose concentration again post-incubation.
Cells were seeded at 1×106 cells per well in 96-well plates. The glycogen content in hepatocytes was determined using the glycogen assay kit (ab65620, Abcam) as per the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using the EZ-ChIP kit (17-371FR, Millipore, Billerica, Massachusetts, USA) to investigate the interaction between TGIF1 and the miR-106b-5p promoter. Mouse hepatocyte suspension or liver tissue samples were crosslinked with 1% formaldehyde for 10 min, followed by quenching with a 0.125 M glycine solution. Tissue samples were then lysed using a homogenizer and sonicated to shear chromatin. Immunoprecipitation was carried out with TGIF1 antibody (1:50, sc-17800 X, Santa Cruz Biotechnology, Shanghai, China) or IgG antibody (1:50, 2729S, Cell Signaling Technology, Danvers, Massachusetts, USA) as a negative control. Eluted products were used for qRT-PCR analysis. Primers are listed in table 1.
Dual-luciferase reporter assay
Synthetic fragments of EGR2 containing the wild-type (WT) or mutated (MUT) binding sites of miR-106b-5p (EGR2-WT, EGR2-MUT) were cloned into the pMIR-report vector (AM5795, Thermo Fisher Scientific). Cells were co-transfected with EGR2-WT or EGR2-MUT constructs alongside miR-106b-5p mimics or mimic negative control (NC). Similarly, synthetic fragments of the miR-106b-5p promoter sequence containing the WT or MUT binding sites of TGIF1 (pro-miR-WT, pro-miR-MUT) were inserted into the pCMV-report vector (16156, Thermo Fisher Scientific). Cells were co-transfected with pro-miR-WT or pro-miR-MUT constructs with overexpressed TGIF1 (oe-TGIF1) or overexpression negative control (oe-NC). After 48 hours post-transfection, cells were collected and lysed. Luciferase activity was measured using the luciferase assay system (K801-200, BioVision, Mountain View, California, USA).
Statistical analysis
SPSS V.21.0 (IBM, Armonk, New York, USA) and GraphPad Prism V.8.0 software (GraphPad Software, San Diego, California, USA) were used for statistical analysis and data plotting. Measurement data are presented as mean±SD. First, normality and homogeneity of variance tests were conducted, which verified that the data were in normal distribution and homogeneity of variance. Data comparison between two groups was performed using t-test. Data comparison among multiple groups was performed using one-way or two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test for post hoc analysis. P value was obtained via two-sided tests; p<0.05 indicated a statistically significant difference.




Comments (0)