
Remember me
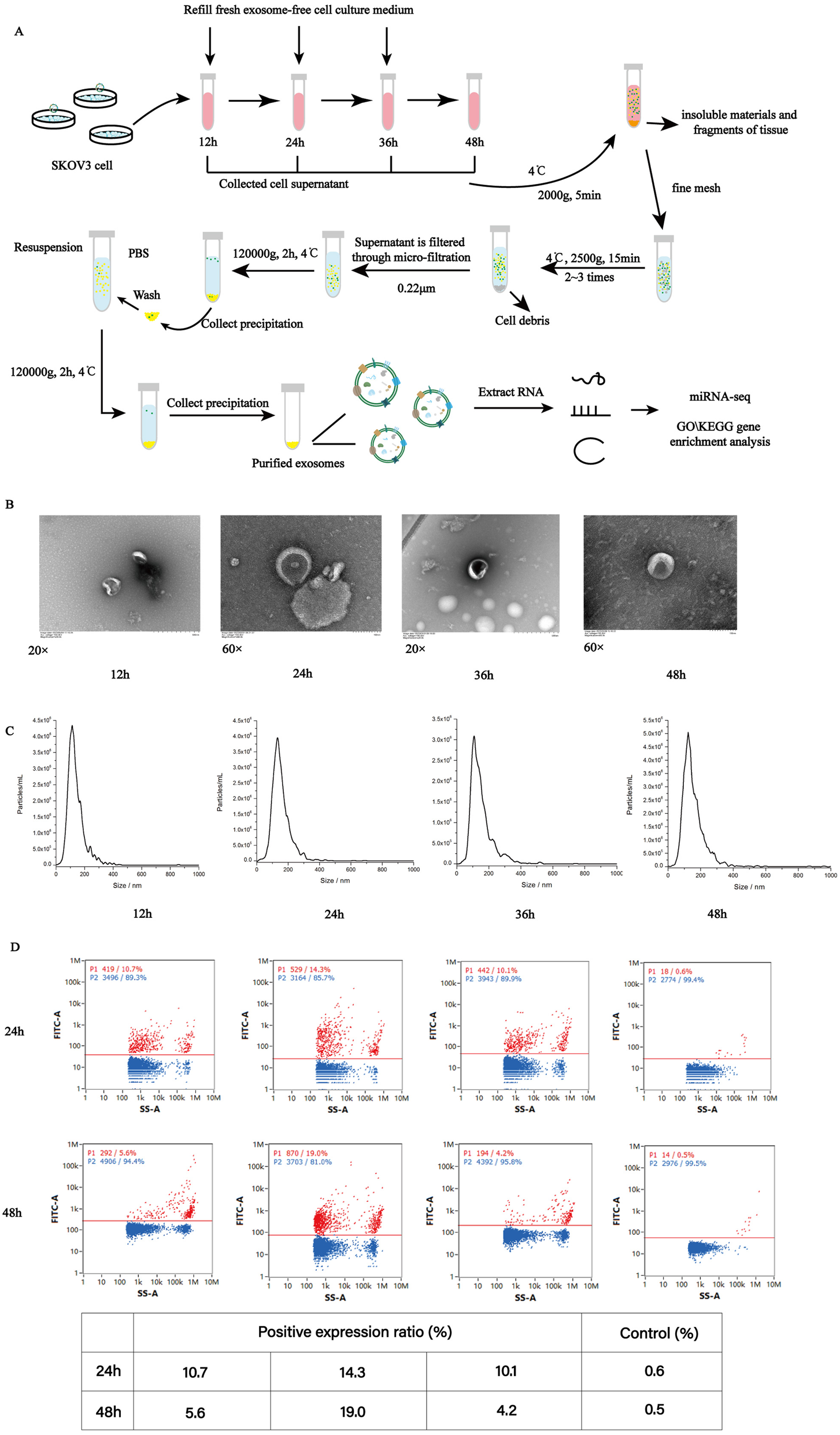





When OC cells passage was stable with good viability and consistent growth status, the experiment was performed. The cell culture medium were collected at different times (12, 24, 36 and 48 h). These samples of exosomes were obtained by a series of differential centrifugation, ultracentrifugation, and filtration steps. The extraction procedure and brief protocol was shown in Fig. 1A.
Fig. 1
Characterization of exosomes from ovarian cancer cells at different culture time points. (A) A schematic diagram illustrating the experimental design from sample collection till sequencing. (B) TEM image of the exosomes. Electron microscopy allowed visualizing membrane-bound nano vesicles sized ~ 100 nm. Scale bar = 100 nm. (C) Analysis of exosomes with NanoSight LM10-HS instrument. (D) Exosome validation by nanoflow fluorescence indicating the CD9, CD63 and CD81 protein markers for exosomes
The examination with a TEM indicated that exosomes from four groups showed characteristic round- or cup-shaped morphology and dimension (Fig. 1B). The NTA indicated that average sizes of exosomes from the different time pionts (12, 24, 36 and 48 h) were 122.3, 139.8, 131.8 and 134.9 nm, respectively (Fig. 1C). Moreover, it was found that the number of exosomes per million of cells at the each time point (12, 24, 36, and 48 h) were 8.2 × 108, 2.3 × 1010, 1.5 × 108 and 4 × 1010, respectively, showing the obvious differences in the rate of secreting exosomes.
Furthermore, exosomes were validated by nanoflow fluorescence detection indicating the markers (CD9, CD63 and CD81) of exosomes (Fig. 1D), which based on the positive expression results of different antibodies on the exosomes surface. All these results indicated that exosomes were isolated successfully and suitable for further investigation.
Identification of exosomal miRNAsPrevious studies indicated that exosomal miRNAs play important roles in remodeling CM [16, 17], so we tried to investigate the miRNAs cargo of exosomes from the four groups. A total of 12 small RNA libraries (T12-a, T12-b, T12-c; T24-a, T24-b, T24-c; T36-a, T36-b, T36-c; T48-a, T48-b, T48-c) were constructed for miRNA-seq with three biological replicates for each group.
Using Illumina HiSeq 2500, > 298.1 million reads were produced, corresponding to ~ 24.8 million sequence reads per library, and the clean reads account for ~ 95.6%. Hierarchical clustering heatmap analyses revealed that the global expression pattern of miRNAs differed obviously among different groups (Fig. 2A).
Fig. 2
Identification of exosomal miRNAs by miRNA sequencing. (A) Hierarchical clustering heatmap analysis for expression pattern of miRNAs from the four (T12, T24, T36 and T48) groups. (B) Venn diagrams of the identified miRNAs from the four (T12, T24, T36 and T48) groups. (C) The miRNA expression were determined by high-throughput sequencing. Transcripts per million (TPM) per million mapped reads was used to calculate the expression levels of miRNAs. (D) The miRNA expression levels of target genes were determined by RT-qPCR and normalized against U6 expression level. Statistical analysis was performed by Student’s t-test and data are presented at the mean ± standard deviation of three replicates. *P < 0.05, **P < 0.01. RT-qPCR, reverse transcription quantitative real-time polymerase chain reaction
Subsequently, these clean reads were matched to Rfam and human reference genome (Ensembl release 102 GRCh38), and the filtered reads were mapped to miRBase. The read counts were normalized to TPM and 131 miRNAs were identified successfully in all samples (Supplementary Table S1). Moreover, 41, 115, 63 and 24 miRNAs were found in T12, T24, T36 and T48 samples respectively, accounting for 31.3%, 87.8%, 48.1% and 18.3% of the all miRNAs identified (Fig. 2B).
RT-qPCR validationTo verify the miRNA-seq results two miRNAs (let-7d-5p and miR-16-5p) were randomly selected from these identified miRNAs and analyzed by RT-qPCR. It showed that the RT-qPCR expression results were highly correlated with miRNA-seq results (Fig. 2C, D). Therefore, the miRNA-seq data were reliable and suitable for further analysis.
Functional analyses of target genes of common exosomal miRNAsInterestingly, only 15 overlapped miRNAs were found in four groups, including let-7c-5p, let-7a-5p, let-7b-5p, let-7f-5p, let-7i-5p, let-7 g-5p, let-7e-5p, miR-122-5p, miR-1246, miR-16-5p, miR-155-5p, miR-423-5p, miR-1290, miR-3960 and miR-181b-5p, and accounted for 11.4% of the all miRNAs identified.
Generally, the miRNAs could regulate the gene expression by binding to complementary target sites in the mRNAs of their target genes. Using the miRanda algorithm on human miRNA and transcript sequences of miRBase and TargetScan, 398 target genes were found, suggesting that the overlapped miRNAs have the capacity to regulate the expression levels of multiple genes.
To identify the pathways associated with these target genes, GO enrichment analysis was conducted, and 94 biological pathways were identified (P < 0.01, FDR < 0.05) (Fig. 3A; Supplementary Table S2). It was found that a large number of enriched terms were associated with signaling, kinase and cell proliferation, including “positive regulation of TOR signaling” (GO:0032008), “positive regulation of Wnt signaling pathway” (GO:0030177), “cell-cell signaling by Wnt” (GO:0198738), “positive regulation of kinase activity” (GO:0033674), “positive regulation of protein kinase activity” (GO:0045860), “regulation of protein serine/threonine kinase activity” (GO:0071900), “positive regulation of protein serine/threonine kinase activity” (GO:0071902), “positive regulation of cell growth” (GO:0030307), “mitotic cell cycle phase transition” (GO:0044772), “regulation of cell growth” (GO:0001558) and “cell growth” (GO:0016049).
Fig. 3
Functional analyses for the target gene of common miRNAs. (A) and (B) GO and KEGG analyses for target genes of overlapped miRNA in T12, T24, T36 and T48 groups. miRNA, microRNA; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. The rich factor reveals the enrichment degree of terms, while vertical axis indicates the names of the enriched terms. The area of the node shows the number of genes, while the p-value is demonstrated by a color scale with the statistical significance increasing from green to red
In addition, KEGG enrichment analysis was performed for these target genes, and 21 functional terms were obtained (P < 0.01, FDR < 0.05) (Fig. 3B; Supplementary Table S3). It indicated that multiple enriched terms were associated with signaling and cancer, including “mTOR signaling pathway” (ID: hsa04150), “IL-17 signaling pathway” (ID: hsa04657), “TNF signaling pathway” (ID: hsa04668), “Signaling pathways regulating pluripotency of stem cells” (ID: hsa04550), “AGE-RAGE signaling pathway in diabetic complications” (ID: hsa04933), “PI3K-Akt signaling pathway” (ID: hsa04151), “MAPK signaling pathway” (ID: hsa04010), “Breast cancer” (ID: hsa05224), “Gastric cancer” (ID: hsa05226), “MicroRNAs in cancer” (ID: hsa05206), “Acute myeloid leukemia” (ID: hsa05221). The results of GO and KEGG analyses suggesting that exosomal miRNAs of OC play important roles in regulating signaling and cancer development.
Functional analyses of target genes of specific exosomal miRNAsIt indicated that 3, 57, 10 and 3 specific miRNAs were also found in T12, T24, T36 and T48 groups (Fig. 2B; Table 1), respectively.
Table 1 The specific exosomal miRNAs in four groupsThere were 199 putative target genes of the specific miRNAs in T12 group, and the KEGG analyses were performed. The results indicated that 34 terms were enriched (P < 0.05, FDR < 0.65) (Fig. 4A; Supplementary Table S4), and the most of terms were associated with cancer and signaling, including “FoxO signaling pathway” (ID: hsa04068), “Colorectal cancer” (ID: hsa05210), Hepatocellular carcinoma” (ID: hsa05225), “Breast cancer” (ID: hsa05224), “MAPK signaling pathway” (ID: hsa04010), “PI3K-Akt signaling pathway” (ID: hsa04151), “Proteoglycans in cancer” (ID: hsa05205), “Signaling pathways regulating pluripotency of stem cells” (ID: hsa04550), “Gastric cancer” (ID: hsa05226), “Basal cell carcinoma” (ID: hsa05217) and “TGF-beta signaling pathway” (ID: hsa04350), “Non-small cell lung cancer”(hsa05223), “Pancreatic cancer”(hsa05212), “Ras signaling pathway”(hsa04014), “ErbB signaling pathway”(hsa04012),”Micro-RNAs in cancer”(hsa05206), “Estrogen signaling pathway”(hsa04915) “GnRH signaling pathway”(hsa04912), “mTOR signaling pathway”(hsa04150), “Hippo signaling pathway”(hsa04390) and “Endometrial cancer”(hsa05213).
Fig. 4
Functional analyses for the target gene of specific miRNAs. (A), (B), (C) and (D) KEGG analyses for target genes of specific miRNAs in T12, T24, T36 and T48 groups. miRNA, microRNA; KEGG, Kyoto Encyclopedia of Genes and Genomes. The rich factor indicates the enrichment degree of terms, while vertical axis shows the names of the enriched terms. The area of the node represents the number of genes, and the p-value is shown by a color scale with the statistical significance increasing from green to red
There were 1601 putative target genes of the specific miRNAs in T24 group, and the KEGG analyses indicated that 92 terms were enriched (P < 0.05, FDR < 0.65) (Fig. 4B; Supplementary Table S5). It was found that the most of terms were associated with cancer and signaling, including “MAPK signaling pathway” (ID: hsa04010), “Renal cell carcinoma” (ID: hsa05211), “PI3K-Akt signaling pathway” (ID: hsa04151), “mTOR signaling pathway” (ID: hsa04150), “MicroRNAs in cancer” (ID: hsa05206), “Neurotrophin signaling pathway” (ID: hsa04722), “Relaxin signaling pathway” (ID: hsa04926), “Transcriptional misregulation in cancer” (ID: hsa05202), “FoxO signaling pathway” (ID: hsa04068), “Small cell lung cancer” (ID: hsa05222), “Proteoglycans in cancer” (ID: hsa05205), “TNF signaling pathway” (ID: hsa04668), “Colorectal cancer” (ID: hsa05210), “p53 signaling pathway” (ID: hsa04115), “Prostate cancer” (ID: hsa05215), “Ras signaling pathway” (ID: hsa04014), “Wnt signaling pathway” (ID: hsa04310), “Bladder cancer” (ID: hsa05219), “Non-small cell lung cancer”(hsa05223), “Endometrial cancer”(hsa05213), “TGF-beta signaling pathway”(hsa04350), “Rap1 signaling pathway”(hsa04015) and other related pathways. Moreover, the “Circadian rhythm” (ID: hsa04710) was also identified.
There were 314 putative target genes of the specific miRNAs in T36 group, and KEGG analyses indicated that 31 terms were identified (P < 0.05, FDR < 0.65) (Fig. 4C; Supplementary Table S6). Some enriched terms were associated with cancer and signaling, including “MAPK signaling pathway” (ID: hsa04010), “Adrenergic signaling in cardiomyocytes” (ID: hsa04261), “Transcriptional misregulation in cancer” (ID: hsa05202) “Renal cell carcinoma” (ID: hsa05211), “Ras signaling pathway” (ID: hsa04014) “mTOR signaling pathway” (ID: hsa04150) “Adherens junction” (ID: hsa04520) “Melanoma” (ID: hsa05218) and“Circadian entrainment” (ID: hsa04713).
There were 171 putative target genes of the specific miRNAs in T48 group, and the KEGG analyses were performed. The results indicated that 12 terms were enriched (P < 0.05, FDR < 0.65) (Fig. 4D; Supplementary Table S7), and most of these terms were associated with cancer and immune signaling, including “Retrograde endocannabinoid signaling” (ID: hsa04723), “Efferocytosis” (ID: hsa04148), “Fc gamma R-mediated phagocytosis” (ID: hsa04666) and “Ferroptosis” (ID: hsa04216).
Validation of the regulation between miRNAs and target geneTo further validate the prediction results of the miRNAs, a dual-luciferase assay was performed for verifying the regulative interaction between miR-320a-3p and its potential target gene (DEC2), which play key roles in regulating cell proliferation, apoptosis, circadian rhythm (CR) and epithelial-to-mesenchymal transition (EMT) of tumor cells [18]. The result showed that miR-320a-3p binding significantly reduced luciferase activity for the Wt target gene, whereas binding to the mutant site had no effect on luciferase activity (Fig. 5). Together, these results suggest that exosomal miRNAs have the potential regulating gene expression in cells.
Fig. 5
The interaction between exsomal miR-320a-3p and its potential target gene (DEC2) was validated by the dual luciferase activity assay. DEC2, differentially expressed in chondrocytes protein 2; ***P < 0.001
Comments (0)