Study design
This is an open-label, pragmatic post-marketing study. The objective of this study was to evaluate the safety and immunogenicity of a quadrivalent influenza vaccine, with the safety endpoints of the incidence of adverse events and serious adverse events, with immunogenicity endpoints as seroconversion rate, seroprotection rate, geometric mean titer (GMT) and geometric mean increase (GMI) of HI antibodies 30 days post-immunization.
This study was carried out in Shandong Province, China, and conducted by the Shandong Center for Disease Control and Prevention (CDC). Before initiation of the study, the protocol, informed consent form (ICF) and other information provided to recipients had been reviewed and approved by the Preventive Medical Ethical Committee of Shandong CDC (No. 2021-70).
Study population
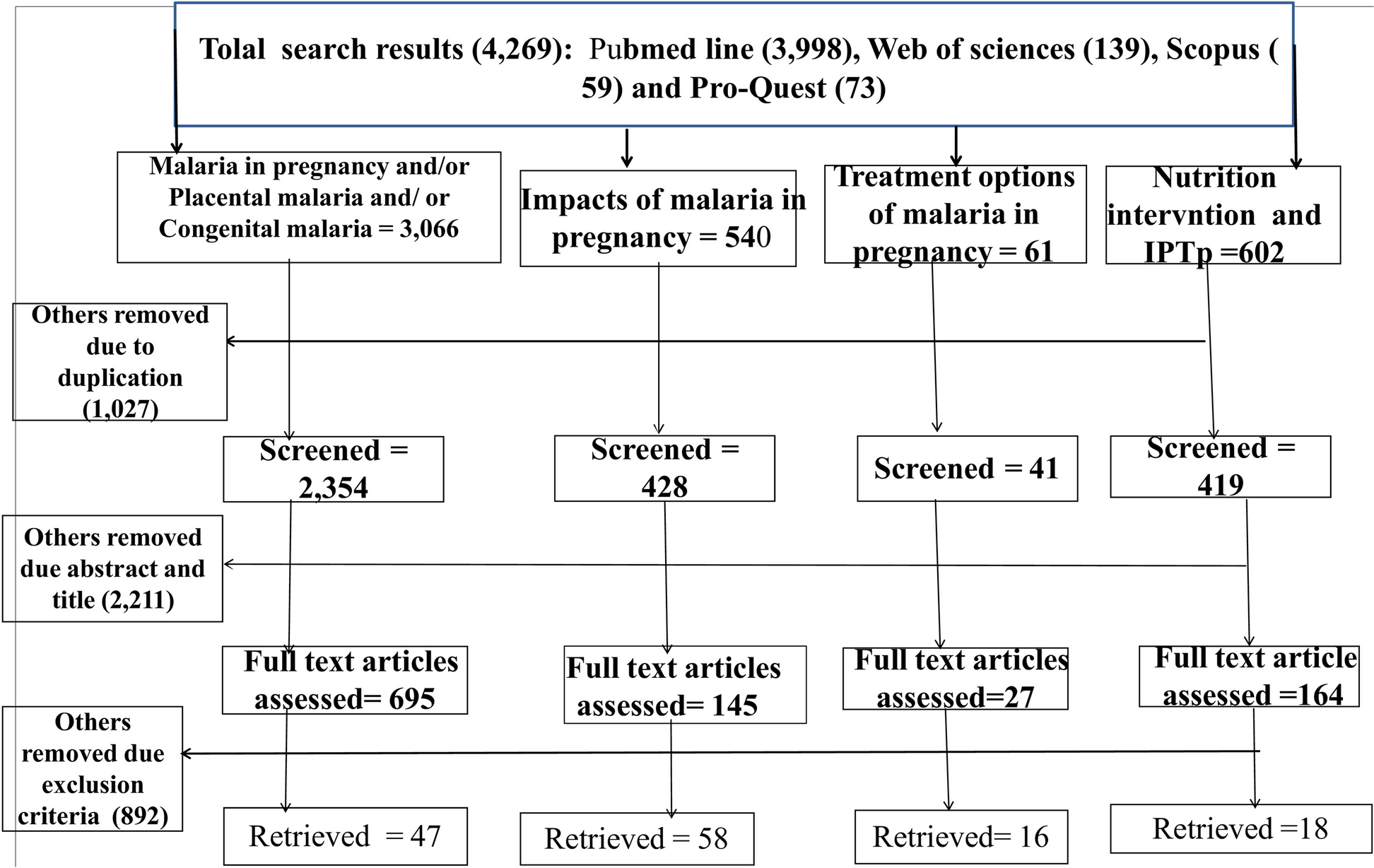
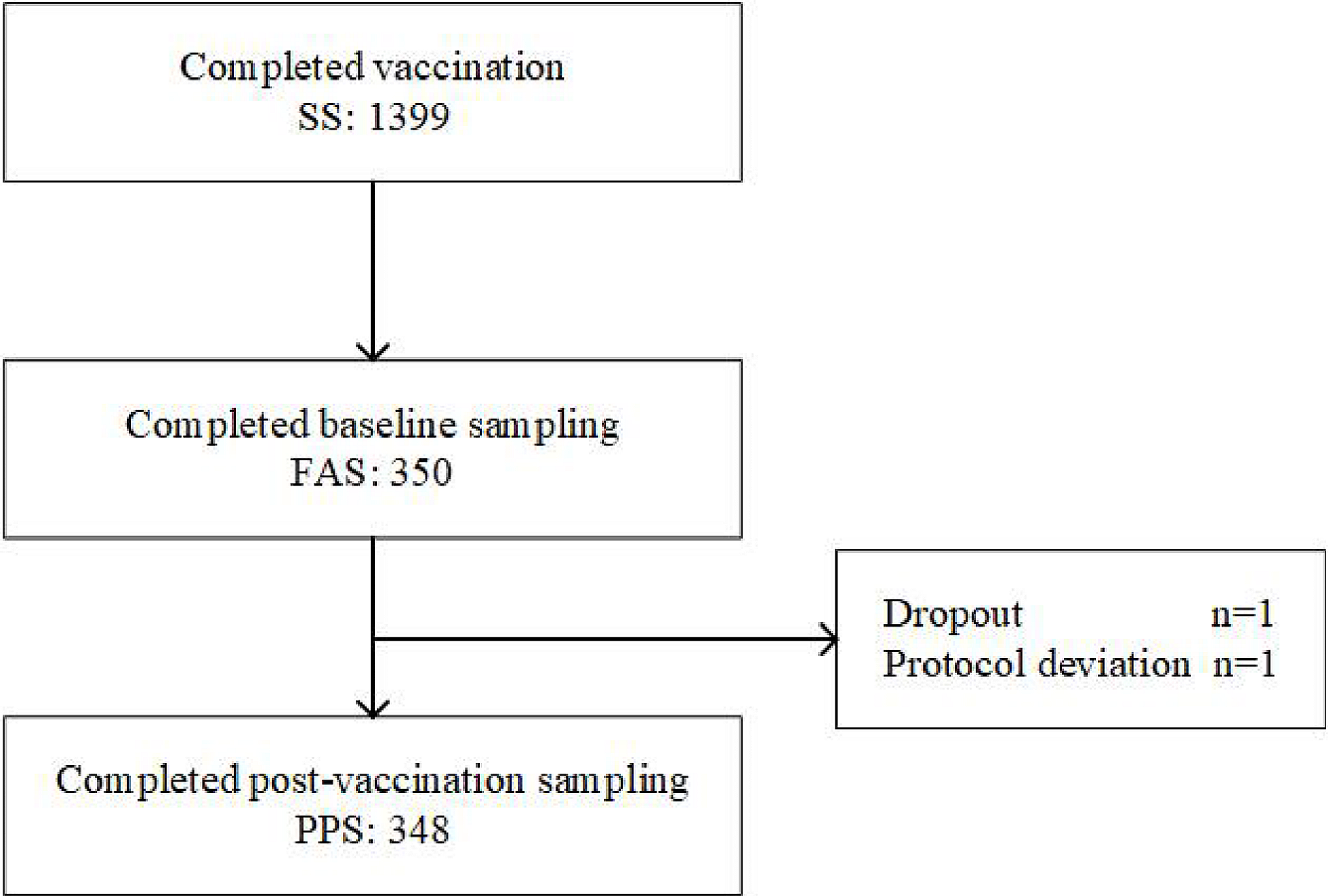
This study enrolled 1399 elderly subjects aged ≥ 60 years, without contraindications noted in the package insert of quadrivalent influenza vaccine. No rigorous physical or laboratory tests were conducted during the screening, because subjects in poor health condition were acceptable for this pragmatic study.
Study vaccine
All screening-eligible subjects received one dose of quadrivalent influenza vaccine at the lateral deltoid muscle of the upper arm. The vaccine used in this study was a commercially available quadrivalent influenza split-virion vaccine produced by Hualan Bio that has been approved in China, that contains no adjuvant and 15 µg hemagglutinin per strain including A/H1N1, A/H3N2, B/Victoria and B/Yamagata. Prefilled syringes with 16 ± 1 mm length needles were used for vaccination, with 2/3 length of the needles injected in the lateral deltoid muscle of the upper arm. Batch No.: 202107B054. Stored and transported in 2 ~ 8℃ condition.
Safety assessment
All vaccinated subjects were observed on-site for 30 min to assess immediate local and systemic adverse events, after which they were followed for 30 days for adverse event collection by recording on a contact card. Long-term safety observations were conducted within 31–180 days with a combination of methods of active monthly follow-up and self-reporting by subjects to collect serious adverse event (SAE) data.
Causality between adverse events and vaccination was analyzed in 5 degrees as: definitely-related, probably-related, possibly-related, likely-unrelated, and definitely-unrelated. Vaccination-related adverse events, including definitely-related, probably-related and possibly-related events, were referred to as adverse reactions. The severity of adverse events was categorized following the Guidelines for the classification of adverse events in clinical trials of preventive vaccines issued by the National Medical Products Administration (NMPA) in 2019 [21]. The collected adverse events were coded according to the Medical Dictionary for Regulatory Activities (MedDRA) and statistically analyzed for incidence and severity.
Immunogenicity assessment
This study evaluated immunogenicity in a subgroup, endpoints of which included seroconversion rate and seroprotection rate of each type of HI antibody elicited by quadrivalent influenza vaccine in the elderly aged ≥ 60 years. Sample size of immunogenicity subgroup subjects was calculated with Confidence Interval of one Proportion method by using PASS 15.0. Assuming that the seroconversion rates of all types of HI antibodies exceeded 55%, the two-side confidence level 1-α = 0.95, the width of the confidence interval was 0.12, and the dropout rate was estimated as 20%, at least 347 subjects should undergo immunogenicity assessment. As a result, 350 subjects (25% of 1400), assigned randomly when enrolled, underwent venous blood sampling pre-vaccination and at 30 days after vaccination, to detect antibody titers. Immunogenicity evaluation was based on antibody titers against each subtype of influenza virus by micro-HI assay with serum separated from collected blood samples.
When statistically analyzed, referring to the NMPA Technical Guidelines for Clinical Research of Seasonal Influenza Virus Vaccine (Exposure Draft) [22], FDA Clinical Data Needed to Support the Licensure of Seasonal Inactivated Influenza Vaccines [17], and the EMEA Note for Guidance on Harmonisation of Requirements for Influenza Vaccines [16], seroprotection was defined as an HI antibody titer ≥ 1:40, and seroconversion was defined as an HI antibody titer change to ≥ 1:40 post-vaccination from baseline < 1:10 or a ≥ 4-fold increase in HI antibody titer post-vaccination from baseline ≥ 1:10. When the antibody titer was < 1:10, a titer of 1:5 was carried forward to calculate the GMT.
According to these guidance issued by NMPA, FDA and EMEA, quadrivalent influenza vaccines could be considered to have favorable immunogenicity among populations aged ≥ 60 years if at 30 days post-vaccination (1) The lower bound of the two-sided 95% confidence interval (CI) for the percentage of subjects achieving seroconversion for HI antibodies meet or exceed 30%; (2) The lower bound of the two-sided 95% CI for the percentage of subjects achieving an HI antibody titer ≥ 1:40 meet or exceed 60%; (3) The lower bound of the two-sided 95% CI for GMI > 2.0.
Statistical analysis
We used SAS 9.4 for the statistical analysis of this study. The incidence of adverse events within 0–30 days post-vaccination, as the primary endpoint, and the incidence of SAEs within 0-180 days post-vaccination, as the secondary endpoint, were statistically described along with their Clopper-Pearson two-sided 95% CIs. For estimation of the primary immunogenicity endpoints, immunity assays and statistics were conducted among 350 subjects who were randomly assigned to an immune subset at the time of enrollment. The seroprotection rate and seroconversion rate were estimated, and the corresponding two-sided 95% CIs were derived from the Clopper-Pearson method. The GMTs and GMIs of the HI antibodies against each type of component (H1N1, H3N2, BV and BY) were calculated together with their two-sided 95% CIs.
The safety set (SS) includes data from all vaccinated subjects with at least one safety observation, while data from subjects with protocol violations were not excluded. The full analysis set (FAS) included data from all vaccinated subjects with detectable results from pre- or post-vaccination serum. The per-protocol set (PPS) includes data from subjects who underwent vaccination and blood sampling following predefined protocol requirements, with valid antibody detection results from pre-and post-vaccination serum.
Serological methods
All the serum samples were treated by the National Institutes for Food and Drug Control in strict accordance with regulations and laboratory manuals. The micro-HI test was used to detect HI antibodies.








Comments (0)