Cells, viruses, and experimental animals
African green monkey kidney (Vero) cells (Procell, Wuhan, China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Basal Media, Shanghai, China) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA), and the SP2/0 cells (Keycell, Wuhan, China) were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640; Basal Media, Shanghai, China) containing 20% FBS.
PEDV CH/SX/2016 strain (GenBank accession no. MT787025.1) was isolated and maintained in State Key Laboratory for Animal Disease Control and Prevention in Lanzhou, China. The cell culture supernatants of porcine reproductive and respiratory syndrome virus (PRRSV), porcine pseudorabies virus (PRV), porcine rotavirus (PoRV), and porcine circovirus (PCV) were provided by State Key Laboratory for Animal Disease Control and Prevention in Lanzhou, China.
BALB/c mice and female rabbits were obtained from Lanzhou Veterinary Research Institute, China Agricultural Science Academy. Weaned piglets were purchased from the Kangzhiyuan Planting Farmers’ Specialized Cooperative in Linxia.
Preparation of PEDV N protein
Using RT-PCR (with primers F:5′- AAATGGGTCGGGATCCGATGGCTTCTGTCAGCTTTCAGGA-3′ (contains a BamHI digestible site) and R: 5′- GGTGGTGGTGCTCGAGATTTCCTGTATCGAAGATCTCGTTGATAATCT-3′ (attached to a XhoI site)) the N protein from the PEDV CH/SX/2016 strain was amplified and subcloned into the pET-21b ( +) (Merck KGaA, Darmstadt, Germany). The pET-21b-PEDV-N positive plasmid was transformed into Escherichia coli BL21 (DE3) (TransGen Biotech, Beijing, China) competent cells. Then, after induction with 1 mM isopropyl β-D-thiogalactoside (IPTG) (Biotopped, Beijing, China) at 37 °C for 6 h, purification was performed using Ni–NTA resin (TransGen Biotech, Beijing, China). Lastly, identification was performed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis.
Preparation and characterization of PAb and MAb against PEDV N
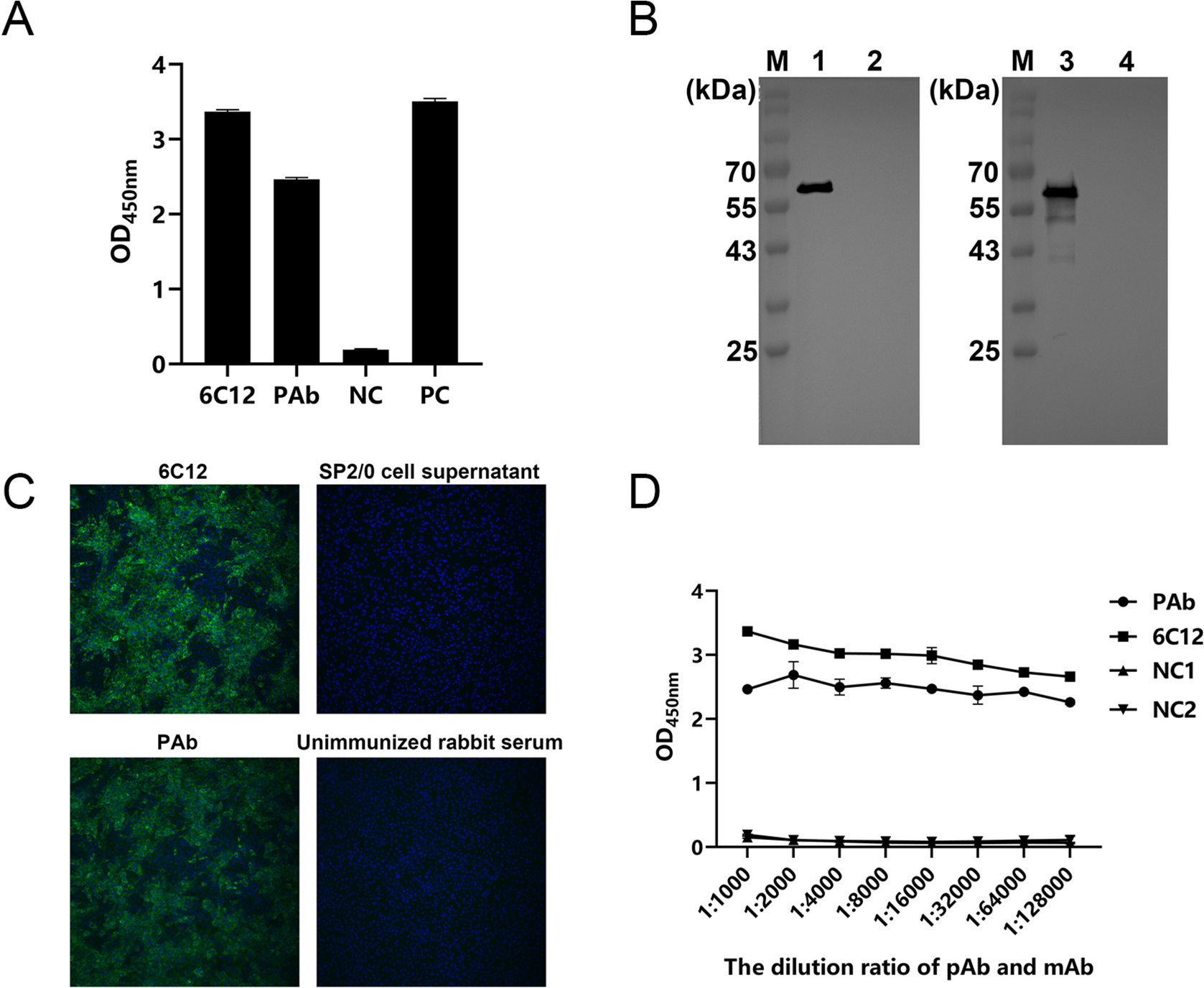
Immunized 6–8-week-old BALB/c female mice (50 µg/mouse) and 8–12-week-old female rabbits (300 µg/rabbit) were used as the study animals. In the first immunization, the purified rN protein was emulsified with an equal volume of Freund’s complete adjuvant (Sigma-Aldrich, St. Louis, MO, USA) and injected subcutaneously into the back of the mice and rabbits. Subsequent immunizations were emulsified with equal volumes of Freund’s incomplete (Sigma-Aldrich, St. Louis, MO, USA) adjuvant. Immunizations were performed every 2 weeks, and blood samples were collected before each immunization. The titer was tested using an ELISA kit coated with N protein. For mice, after three immunizations, when the titer exceeded 1 × 104, a booster immunization was administered via intraperitoneal injection. Three to five days after immunization, peritoneal washes from blank mice were collected as a source of feeder cells to facilitate hybridoma cell growth. Induced fusion of SP2/0 cells and spleen cells from immunized mice into hybridoma cells was obtained using PEG1450 (Sigma-Aldrich, St. Louis, MO, USA) and cultured using HAT/HT selection medium (Sigma-Aldrich, St. Louis, MO, USA). Indirect ELISA was used to detect the supernatant of the cell cultures, followed by screening for positive hybridoma cells and three rounds of subclone culturing on positive hybridoma cells to obtain hybridoma cells that stably secreted MAb against the PEDV N protein. Female BALB/c mice aged 8–10 weeks were sensitized via intraperitoneal injection of an incomplete adjuvant. After 1 week, 1 × 107 hybridoma cells were injected into the peritoneal cavity of each mouse to prepare the ascitic fluid. The MAb subtype was identified using a mouse MAb IgG subclass identification kit (Biodragon, Suzhou, China). Serum was collected from rabbits that had demonstrated high levels of potency. Subsequently, the rabbit serum and mouse ascites were purified using protein G resin (Biodragon, Suzhou, China) and the antibody titers were determined by indirect ELISA. The antibodies were further validated by indirect immunofluorescent assay (IFA) and western blotting, and MAb were labeled using HRP labeling reagent kits (Biodragon, Suzhou, China).
Enzyme-linked immunosorbent assay
The purified rN protein was diluted to 2 ng/µL in phosphate buffer saline (PBS) and coated onto ELISA plates overnight at 4 °C. The plates were washed four times with phosphate buffer saline containing 0.05% Tween-20 (PBST) and blocked with blocking buffer (Thermo Fisher Scientific, Waltham, MA, USA) for 2 h at 37 °C. For positive selection, 50 µL of hybridoma cell culture supernatant was added to each well and incubated for 60 min at 37 °C. After four washes, 50 µL of 1:10,000 dilution of HRP-labeled goat anti-mouse IgG secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA) was added and incubated for 60 min at 37 °C. After the same four washes, 50 µL of 3,3’,5,5’-tetramethylbenzidine (TMB) (Biodragon, Suzhou, China) was added, and the plate was incubated for 15 min in the dark. Next, 50 µL of 2 M sulfuric acid was added to terminate the reaction, followed by reading of the absorbance at 450 nm (OD450 nm). Mouse serum was used as the positive control, while the negative control was SP2/0 cell culture supernatant. A sample was considered positive if the OD450 nm value divided by that of the negative control was greater than 2.
To test the titers of PAb and MAb, antibodies were diluted from 1:1000 to 1:12,800 for incubation. The remaining steps were the same as those described above.
Indirect immunofluorescence assay
Vero cells grown in 96-well plates were infected with PEDV and cultured for 24 h at 37 °C in a CO2 incubator. Cells were washed two times with PBS, fixed with 4% paraformaldehyde for 15 min at 37 °C, washed, and permeabilized with 0.1% triton X-100 (Solarbio, Beijing, China) for 15 min. After washing, the cells were blocked cells with 5% bovine serum albumin (BSA) for 30 min at 37 °C, followed by washing three times. Then, the cells were incubated with a 1:1000 dilution of MAb or PAb for 1 h at 37 °C. After three PBS washes, a 1:500 dilution of fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse (or anti-rabbit) IgG (Jackson Immuno Research, West Grove, PA, USA) was added, followed by incubation at 37 °C away from light for 1 h. Following three more washes, the cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) (Jackson Immuno Research, West Grove, PA, USA) at a 1:1000 dilution for 10 min protected in the dark. Finally, the cells were observed under a fluorescence microscope.
Western blotting
The treated protein samples were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. After being closed with 5% skim milk diluted in PBST for 2 h at 37 °C, the membranes were incubated in antibody overnight at 4 °C. After washing four times with PBST, the cells were incubated with an HRP-labeled goat anti-mouse (or anti-rabbit) IgG secondary antibody at 37 °C for 1 h. After another four washes, the membrane was exposed using electrochemiluminescence (ECL).
To validate the purified rN protein, anti-His mouse monoclonal antibody (TransGen Biotech, Beijing, China) was used as the detection antibody to confirm protein expression. The protein samples were detected as either PEDV-infected Vero cell lysates or normal Vero cell lysates, and the antibodies were detected as MAb or PAb to confirm the application of the antibodies in western blotting analysis.
Establishment of DAS‑qELISA
In DAS-qELISA, rabbit PAb was used as the capture antibody and HRP-labeled MAb (6C12). The optimal concentration for capturing and detecting antibodies was determined using checkerboard titration. The purified PAb was diluted (0.5, 1.0, 2.0, and 4.0 µg/mL) using carbonate-bicarbonate buffer (Sigma-Aldrich, St. Louis, MO, USA), adding 50 µL laterally into ELISA plates and incubated overnight at 4 °C. The plates were then washed four times with PBST, followed by incubation with 5% skim milk diluted in PBST for 2 h at 37 °C. After washing, 100 µL was added to each of the PEDV infection and normal Vero culture supernatant and incubated for 1 h at 37 ℃. After washing four times, HRP-labeled MAb was diluted at 1:500, 1:1000, 1:2000, 1:4000, 1:6000, and 1:8000 with PBST and vertically added to the wells (100 µL/well) before incubating for 1 h at 37 °C. Following another round of washing, 50 µL of TMB was added to each well. The reaction was terminated using 50 µL of 2 M sulfuric acid after color development for 10 min at 37 °C, followed by measurement at OD450 nm. The optimal capture and detection antibody concentrations were determined using the highest positive/negative (P/N) ratio.
Based on the determined antibodies concentration, the optimal coating conditions were determined by coating at 37 ℃ for 1 h, 37 ℃ for 1 h + 4 ℃ for 12 h, 37 ℃ for 2 h, 37 ℃ for 2 h + 4 ℃ 12 h, and 4 ℃ for 12 h, respectively. This was followed by incubation with 1.5%, 3.0%, and 5.0% skim milk and BSA at 37 ℃ for 60 min, 90 min, and 120 min, respectively, to determine the optimal blocking conditions. Similarly, the samples were then incubated with HRP-labeled antibody at 37 ℃ for 30 min, 60 min, 90 min, and 120 min to determine the optimal incubation time. Finally, the samples were incubated with TMB at 37 ℃ for 5 min, 10 min, 15 min, and 20 min, respectively, to determine the optimal conditions.
Cutoff value of DAS‑qELISA
PEDV-negative anal swabs were collected from 30 healthy piglets, diluted with 1 mL of PBS, and vortexed for 30 s. The supernatant was collected after centrifugation at 13,000 × g for 5 min. Each sample was analyzed three times, detected using the established DAS-qELISA, and measured at OD450 nm. The average value and standard deviation (SD) of the OD450 nm values were calculated, and the cutoff value was determined as equal to the sum of the average value and three times the SD. When the OD450 nm value of the test sample was greater than the cutoff value and the sample-to-negative (S/N) ratio was greater than 2, it was deemed to be positive; otherwise, it was considered negative.
Sensitivity, specificity, and reproducibility
Purified rN protein was diluted with PBS from 200 to 0.012 ng/mL, with PBS used as the negative control. The 50% tissue culture infective dose (TCID50) of the PEDV cell culture was measured as 105.13 TCID50/mL, and the PEDV cell culture was diluted with DMEM from 1:16 to 1:4096. The normal Vero supernatant was used as the negative control. The absorbance was measured at OD450 nm and the S/N value was analyzed to determine the sensitivity of DAS-qELISA.
Next, the specificity of DAS-qELISA was evaluated using PRRSV, PoRV, PRV, porcine deltacoronavirus (PDCoV), and PCV. PEDV-infected cell culture supernatants were used as positive controls and PBS was used as a negative control.
To test the intra-assay reproducibility, four PEDV cell culture supernatants and normal cell culture supernatants were assayed four times. Four batches of DAS-qELISA were used to confirm inter-assay reproducibility based on the average value, SD, and coefficient of variation (CV) of the absorbance measured at OD450 nm.
Compliance rate test
To determine the detection effect of DAS-qELISA on clinical samples, an animal test was conducted on twelve one-day-old piglets by oral gavage with the PEDV (CH/SX/2016) strain. DMEM was used as a negative control for six piglets taken orally, with anal swabs collected daily. The collected anal swabs were then diluted with 1 mL of PBS and vortexed for 30 s. The resulting supernatant was collected by centrifugation at 13,000 × g for 5 min. Then, 300 µL of extracted RNA was taken for RT-PCR detection and 100 µL was used for DAS-qELISA detection, and the consistency of the two detection methods was compared based on the results. In addition, 35 samples were randomly selected for the PED Ag Rapid Test Kit (JNT, Beijing, China), and the results were compared with those of the DAS-qELISA.








Comments (0)