
Remember me
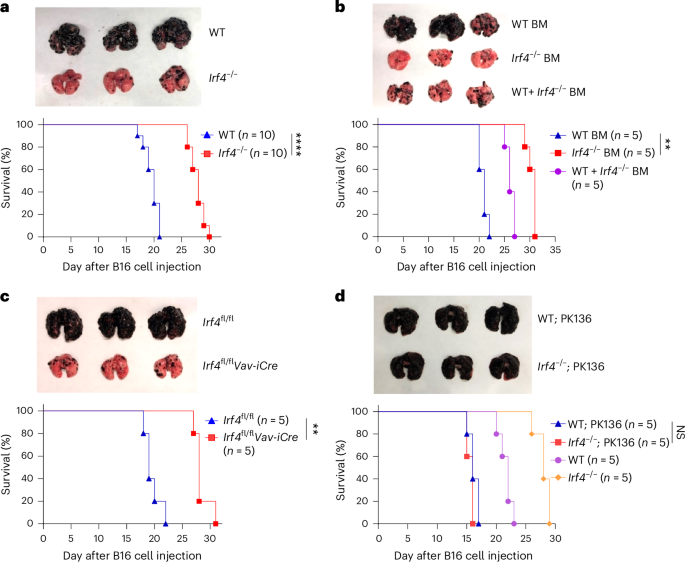


Experiments in mice were performed in accordance with the animal care guidelines of the European Union ARRIVE and French laws and were validated by the local animal ethics evaluation committees CECAPP and the French Ministry of research under approbation numbers CECAPP CLB 2017‐017 and APAFIS 18685. No animal developed intestinal inflammation/tumors reaching a state that could alter their daily lives in any manner. For human data reanalysis, ethics statements were reported in the original published articles47,48,49.
MiceIl17a‐cre mice were provided by G. Stockinger (The Francis Crick Institute)14, and Vil1-creErt2 mice were provided by S. Robine (Institut Pasteur)50. Stopfl/fl-Tgfbr1-CA mice were generated as previously described16. Tgfbr2fl/fl mice were provided by S. Karlsson (Lund university)15, Rosa26‐stopfl/fl-yfp mice were provided by F. Constantini (Columbia University)51, Trim3fl/fl mice were provided by R. Losson (Ilkirch), Smad4fl/fl mice were provided by C. Deng (NIH)52, C.129S6-Tbx21tm1Glm/J (T-BET-KO) mice were provided by L. Glimcher (Havard University) and Tgfb1tm2.1DoeTgfb1fl mice were provided by O. Sansom (Glasgow University)53. Il17a-KO animals were obtained from homozygous Il17a‐cre animals using a knock-in approach after insertion of the construct in both Il17a alleles14. RAG-KO (B6.Cg-Rag2tm1.1Cgn/J) mice (008449, Jackson Laboratory) and C57BL6/J mice (000664, Jackson Laboratory) were purchased from Charles River, and colonies were maintained. Except when mentioned, experiments were performed on 6- to 8-month-old mice. All mice were on a C57BL/6 background, and both sexes were used without differences between males and females being observed except for AOM DSS, in which males were used because they responded more strongly. Littermates without floxed alleles were used as WT control mice. Except for the microbiota analysis, TGFβR-WT and TGFβR-KO animals were cohoused throughout their lifespan. All animals were maintained in specific pathogen‐free animal facilities (Small Animal Platform/Animal Core Facility/Imaging Platform or RAM-ZEFI). Irradiated diet chow (2918 Envigo; A04 and 150 SP-25 Safe Diets) was provided without any alteration in our observations. Animals were housed with enrichment media, including light shed houses and cotton squares, on a 12-h light/12-h dark cycle in a controlled environment (temperature of 22 + 1 °C and hygrometry of 50–60%).
AOM DSS treatmentMice were injected intraperitoneally (i.p.) with AOM (6.25 mg per kg (body weight); 25843-45-2, Sigma-Aldrich), followed 30 days later by a single 5-day period of 2.5% (wt/vol) DSS (TdB) administered in the drinking water. Because TGFβR-KO mice did not regain weight after DSS treatment, no other induction of inflammation was performed, as is the case in the classical colitis-associated carcinogenesis protocol54. Mouse weight was measured every other day, and when an over 20% weight loss was observed, mice were killed. Animals were killed after 80 days, and organs were collected for histological analysis.
Tamoxifen and anti-IFNγ treatmentMice were injected i.p. every day over 5 days with 100 μl of a 10 mg ml–1 tamoxifen solution (T5648, Sigma-Aldrich) dissolved with pure ethanol and diluted in corn oil (C8267, Sigma-Aldrich), allowing Cre activity in the intestine for 60 days50. For anti-IFNγ treatment, mice were injected i.p. every 3 days with 200 µg of either neutralizing anti-IFNγ (XMG1.2, BioXCell) or IgG isotype control (HRPN, BioXCell).
Isolation of intestinal cellsThe small intestine and colon were dissected, and fat was removed. Intestines were longitudinally opened and washed in 1× PBS (14200091, Gibco). Intestines were cut into small pieces and incubated with 5 mM EDTA (EU0084, Euromedex) and 1 mM DTT (D0632, Sigma-Aldrich) at 37 °C. Epithelial cells were then separated from intraepithelial lymphocytes with a 44%/67% Percoll gradient (P1644, Sigma-Aldrich) run for 20 min at 1,300g. Tissues were then digested in RPMI medium (618700044, Gibco) containing 20% fetal bovine serum (FBS; 10437028, Gibco), 100 µg ml–1 DNase I (DN25-IG, Sigma-Aldrich) and 1 mg ml–1 collagenase from Clostridium histolyticum (C2674, Sigma-Aldrich). Intestinal lamina propria lymphocytes were then separated on a 44%/67% Percoll gradient run for 20 min at 1,300g.
Flow cytometry and cell sortingSurface staining was performed using the following fluorescence-conjugated antibodies diluted in 1× PBS (Gibco) containing 2% bovine serum albumin (A7906, Sigma-Aldrich) and 0.1% sodium azide (08591, Sigma-Aldrich): CD45-APC-Cy7 (30-F11, BD Biosciences), TCRβ-PE (H57-597, BD), CD3-BV650 (145-2C11, BD Biosciences), CD4-BV711 (RM4-5, Biolegend), CD8-BV510 (53-6.7, BD Biosciences) and CRTAM (11-5/CRTAM-PE, Biolegend). For intracellular cytokine staining, mouse cells were restimulated ex vivo for 4 h with 500 ng ml–1 PMA (P1585, Sigma-Aldrich) and 500 ng ml–1 ionomycin (I0634, Sigma-Aldrich) in the presence of brefeldin A (BD Biosciences). Cells were stained for surface markers and fixed for 30 min with 1% paraformaldehyde (PFA; 11481745, Fisher Scientific) in 1× PBS (1420091, Gibco) and permeabilized with a Cytofix/Cytoperm kit (554655, BD Biosciences) according to the manufacturer’s protocol. Intracellular staining was performed for IL-17A-AlexaFluor 700 (TC11-18H10, BD Biosciences), IFNγ-APC (XMG1.2, BD Biosciences), GM-CSF-PerCP-Cy5.5 (MP1-22E9, Biolegend), TNF-PE-Cy7 (MP6-XT22, BD Biosciences) and granzyme B-APC (GB11, Invitrogen). For intranuclear staining, cells were treated with a nuclear fixation kit (Ebioscience) before being stained with RORγt-PE-CF594 (Q31-378, BD Biosciences), T-BET-APC (eBio4B10, Ebioscience), granzyme B-APC (GB11, Invitrogen) and KLF6-PE (E-10, Santa Cruz). For p-SMAD2/SMAD3 staining, after a 15-min incubation with 5 ng ml–1 activated recombinant TGFβ1 (240-B010, R&D Systems) at 37 °C, cells were immediately fixed with a Fixation and Permeabilization Buffer kit (00-5523-00, eBioscience) and stained with anti-p-SMAD2/SMAD3 (D27F4, Cell Signaling) detected with a donkey anti-rabbit APC secondary antibody (A31573, Life Technology). All samples were acquired on a BD Fortessa except for samples processed with the CRISPR–Cas9 approach and used for intestinal segment analysis, for which an AURORA Cytek machine was used. Analyses were performed with FlowJo v10.6.1 software (BD Biosciences). For cell sorting, CD4+ T cells were enriched using a CD4+ T Cell Isolation kit (mouse; 130-104-454, Miltenyi Biotec) and labeled with CD4-PE (GK1.5, eBioscience), TCRβ-APC (H57-597, BD Biosciences), CD45-APC-Cy7 (30-F11, BD Biosciences) and DAPI (Eurobio Scientific). All antibodies were used at 1:200 except RORγt-PE-CF594 (used at 1:400) and p-SMAD2/SMAD3 and CRTAM (both used at 1:100). Cells were purified on an Aria II (BD Biosciences) according to Extended Data Fig. 2.
Adoptive T cell transferSorted CD4+TCRβ+NK1.1– cells (1 × 106) isolated from the mLNs of either TGFβR-WT or TGFβR-KO mice were injected intravenously (i.v.) into RAG-KO mice. After electroporation with CRISPR–Cas9 and guide RNA (gRNA), purified YFP+CD4+TCRβ+ cells (1 × 105) from the SILP of TGFβR-KO mice were injected i.v. into C57BL/6 WT mice. Sorted YFP+CD45+CD4+TCRβ+ cells (5 × 104) isolated from the small intestines of either TGFβR-WT or TGFβR-KO mice were injected i.v. into either IECΔTgfb1 or IECWT recipients.
Histology and pathology scoringThe small intestine and colon were fixed in 4% PFA in PBS overnight and maintained in 70% ethanol diluted in distilled water. Samples were embedded in paraffin, sliced into 5-μm-thick sections, mounted and stained with H&E using standard protocols. All microscopy acquisitions were performed on a Zeiss Axio Imager M2 and visualized with a NanoZoomer slide scanner controlled by NDP.view software. Scoring of histopathology was performed blinded using the method described by el Marjou et al.50. Briefly, for all pathologic scoring, the following eight parameters were used: (1) the degree of inflammatory infiltrate in the lamina propria, ranging from 1 to 3; (2) the loss of Goblet cells, ranging from 0 to 2; (3) epithelial hyperplasia, ranging from 0 to 4; (4) cryptitis, ranging from 0 to 2; (5) number of crypt abscesses, ranging from 0 to 3; (6) extent of crypt loss regions, ranging from 0 to 2; (7) mucosal erosion to advanced ulceration, ranging from 0 to 4; and (8) presence of adenoma, ranging from 5 to 6. The severity of the inflammatory changes in the distal colon and duodenum correspond to the addition of the scores reported for each parameter.
Intestinal microbiota analysisMicrobial DNA from 200 mg of fresh stools of 3-month-old TGFβR-WT and TGFβR-KO mice was extracted by GenoScreen. Microbial diversity and composition were determined for each sample by targeting a portion of the ribosomal genes. A 16S rRNA gene fragment comprising V3 and V4 hypervariable regions (16S; 5′-TACGGRAGGCAGCAG-3′ and 5′-CTACCNGGGTATCTAAT-3′) was amplified using an optimized and standardized 16S amplicon library preparation protocol (Metabiote v2.0, GenoScreen). Sequencing was performed using a 250-bp paired-end sequencing protocol on an Illumina MiSeq platform (Illumina) at GenoScreen. Positive (artificial bacteria community comprising eight different bacteria (‘ZymoBIOMICS’)) and negative (sterile water) controls were also included. Raw paired-end reads were processed in a data curation pipeline that included a step to remove low-quality reads (Qiime2 2020.8). The remaining sequences were assigned to samples based on barcode matches, and barcode and primer sequences were then trimmed. The sequences were denoised using the DADA2 method, and reads were classified using the Silva reference database (version 138). The α- and β-diversities were computed. Chao1 and Shannon indexes were calculated to characterize α-diversity, and principal coordinate analyses of the Bray Curtis distance and the unweighted UniFrac distance were performed to assess β-diversity using Qiime2 2020.8. Chao1 and Shannon indexes were calculated to characterize α-diversity.
TH1 cell differentiationCD4+ T cells were purified from mLNs (130-104-454, Miltenyi Biotec.). In total, 2 × 105 cells were activated in a 96-well Nunc plate (Thermo Fisher) in the presence of anti-CD3 (1 μg ml–1; 145-2C11, eBioscience) and anti-CD28 (0.5 μg ml–1; 37.51, eBioscience) in RPMI medium supplemented with 10% FBS. Culture medium was completed with 10 ng ml–1 IL-12 (419-ML-010, R&D Systems) and 20 μg ml–1 anti-IL-4 (BE0045, BioXcell). Cells were cultured at 5% CO2 and 37 °C for 3 days. On average, 70% of the cells expressed IFNγ by 3 days of polarization culture.
Immunofluorescence stainingSamples collected from TGFβR-WT, TGFβR-KO and TGFβR-KO; T-BET-KO mice were fixed in 4% PFA overnight before paraffin inclusion. Four-micron-thick slides were washed twice with PBS, permeabilized with 0.2% Triton X-100 (X100, Sigma-Aldrich) and blocked in PBS with 2% bovine serum albumin (A7906, Sigma-Aldrich) for 1 h at room temperature. Slides were then incubated with primary antibodies overnight at 4 °C. For mouse tissue staining, anti-GFP (A-11122, Invitrogen), CD4-PE (GK1.5 eBioscience) and anti-γH2AX (9718S, Ozyme) were used. Secondary antibodies (goat anti-rabbit AlexaFluor 488; A32731, Invitrogen) were incubated for 1 h at room temperature. For nucleus detection, DAPI (D3571, Invitrogen) was used. Images were then acquired using a Confocal Zeiss 980 microscope and analyzed with ImageJ software.
scRNA-seqAfter tissue dissection and dissociation, fluorescence-activated cell sorting-purified suspended YFP+CD4+ T cells were immediately partitioned into nanoliter-scale Gel Bead-In-Emulsions (GEMs) with a Chromium Single Cell Controller (10x Genomics) at the Cancer Research Center of Lyon (CRCL) Single Cell Platform. Cell encapsulation and barcoding were followed by the standard scRNA-seq protocol, including reverse transcription, amplification and indexing (10x Genomics). Sequencing was performed using a NovaSeq Illumina device (Illumina). Illumina bcl files were base called, demultiplexed and aligned to the mouse mm10 genome using CellRanger software (10x Genomics). The output of CellRanger was used to run the Python package velocyto and produce loom files for each sample with RNA velocity estimations55. Loom files from two independent sequencing runs were imported into R, and single-cell data were analyzed with the ‘Seurat’ package56. The first batch included barcoded TGFβR-KO and TGFβR-WT TH17 cells, whereas the second batch was composed of TGFβR-CA and TGFβR-WT cells. Integration of these two datasets followed the ‘Fast integration using reciprocal PCA (RPCA)’ protocol. After filtering for library size (between 1,000 and 5,000 features per cell) and mitochondrial gene expression (less than 10%), preprocessing was performed using Seurat functions for counts normalization (SCTransform using the ‘glmGamPoi’ method). RPCA integration using 3,000 integration features was followed by dimension reduction with principal component analysis (RunPCA with default parameters), construction of a shared nearest neighbor graph (FindNeighbors using ten dimensions of reduction as input based on an elbow plot of variance captured by each principal component), clustering (FindClusters with a resolution of 0.5) and visualization with the UMAP dimensional reduction technique. Initial marker identification was used to identify and remove γδT cells (based on the expression of ‘Tcrg’ and ‘Trg’ genes) and contaminating epithelial cells. In total, 5,080 cells remained for downstream analysis after this final filtering step (TGFβR-CA = 1,098, TGFβR-WT = 1,007 and TGFβR-KO = 2,975). Markers of each cluster were identified using the Wilcox test option of the FindAllMarkers function, with a logarithmic fold change threshold of 0.25 and adjusted P value under 5%. Known markers were used to identify and relabel the resulting clusters, as described in the text. In addition, the COMET package57 was used to identify and validate surface markers distinct from each cluster. A G2/M cell cycle phase score was calculated using an established list of cell cycle markers25. Regulatory network analysis was performed to identify core TFs orchestrating transcriptional programs within each cluster using the single-cell regulatory network inference and clustering (SCENIC) R package58 (https://github.com/aertslab/SCENIC). SCENIC infers coexpression modules between TF and candidate target genes (that is, regulons) using machine learning. Once all regulons are constructed, SCENIC scores its corresponding activity for each individual cell. We used the dynverse R package to compare across pseudotime algorithms and choose the one best suited for our integrated dataset59. Final trajectory (pseudotime) analyses were performed using the slingshot R package60 To this end, clustering information was extracted from Seurat objects for each individual condition and passed directly to slingshot’s main function. The same ‘granularity’ parameters (that is, Omega = 3) were used for all conditions to ensure comparability. Once pseudotime trajectories were identified, a general additive model was fitted to identify genes whose expression was significantly associated with each trajectory. CellRank was used for directional trajectory analyses61. CellRank combines trajectory inference (pseudotime) with directional information from RNA velocity, automatically predicting initial, intermediate and terminal cell populations.
scATAC-seqCD4+YFP+ T cells were purified from the small intestine, including PP, from TGFβR-WT and TGFβR-KO mice. Cell nuclei were independently prepared and frozen following the recommended conditions for scATAC-seq using the 10x Genomics protocol for library preparation (outsourced with ActiveMotif). Thirty-four-base pair paired-end sequencing reads were generated by Illumina Sequencing using a NextSeq 500. Reads were mapped to the mm10 genome, and peaks were called using CellRanger ATAC software with default parameters (mkfastq and count functions). The Signac package (https://satijalab.org/signac/news/index.html) was used in combination with Seurat for all downstream analyses after Tn5 mapping. Briefly, the pipeline includes the creation of a chromatin assay to which nucleosome signal, transcription start site enrichment and fragment data are consecutively added. Next, latent semantic indexing is performed using the ‘RunTFIDF’ and ‘RunSVD’ functions, followed by clustering and UMAP visualization. Gene activity is inferred using the ‘GeneActivity’ function. Merging of TGFβR-WT and TGFβR-KO samples was performed after selecting common good-quality peaks, resulting in 90,255 features across 3,860 cells (after filtering out ten Tcrg/Trg-expressing γδT cells). The combined dataset was further annotated using label transfer from the scRNA-seq data following Seurat’s recommended protocol. Differential accessibility was performed using the ‘FindAllMarkers’ function on peak assay data. Motif analyses were performed with Signac’s ‘AddMotifs’ and ‘RunChromVAR’ functions.
Single-cell TCR sequencing and repertoire analysisAs with scRNA-seq, fluorescence-activated cell sorting-purified suspended YFP+CD4+ T cells from the small intestine were partitioned into nanoliter-scale GEMs with the Chromium Single Cell Controller (10x Genomics), and gene expression and TCRαβ libraries were prepared using a Chromium Single Cell 5′ Library & Gel Bead kit (10x Genomics), as per the manufacturer’s instructions. Sequencing was performed using a NovaSeq Illumina device (Illumina). Illumina bcl files were base called, demultiplexed and aligned to the mouse mm10 genome using CellRanger version 7.1.0 (10x Genomics) in ‘multi’ mode (gene expression + vdj). Demultiplexed data were loaded into R and analyzed with the ‘scRepertoire’ package version 1.12.0 (https://f1000research.com/articles/9-47/v2). scRepertoire was used to assign clonotypes based on TCR chains, quantify and study clonotype dynamics and integrate with gene expression data in combination with the Seurat package. Clonotypes were called by a combination of CDR3 nucleotide sequence and VDJC gene sequence (CTstrict). Shared clonotypes were defined as clonotypes coming from different cell types and containing the same CDR3 nucleotide and VDJC gene sequences.
ChIPSorted YFP+CD4+ T cells were processed with a CUT&RUN assay kit (Cell Signaling), following the manufacturer’s protocol. Anti-KLF6 (sc-365633, Santa Cruz) was used at 2 μg per sample for ChIP. DNA was then purified using DNA spin columns (14209S, Cell Signaling), and 10 ng of DNA per reaction was used for quantitative real-time PCR using LightCycler 480 SYBR Green Master (4707516001, Roche). The following primer sequences were used: Tbx21 CNS0: forward 5′-CTGGAAAATCAGGCTCACGC-3′ and reverse 5′-ACTTTTCCCAGCTTCGAGGA-3′; Tbx21 intragenic region: forward 5′-CACATGAAGTAGGAAGCGCC-3′ and reverse 5′-GGGGAGAGCTGGTGTTAAGT-3′.
Chromatin accessibilityTbx21 DNA accessibility was tested on sorted YFP+CD4+ T cells using an EpiQuik Chromatin Accessibility Assay kit (EpiGenTek) following the manufacturer’s protocol. Isolated chromatin from 2 × 104 cells was then amplified by quantitative PCR. Quantitative PCR was performed using LightCycler 480 SYBR and the same primer sequences depicted above. Fold enrichment was then calculated by the formula fold enrichment = \(2^}}_-}\,}}_)}\) × 100, where NnseCt refers to nuclease-treated sample cycling threshold (Ct), and no NnseCt refers to the control nontreated sample.
CRISPR–Cas9 deletiongRNAs were purchased from Sigma, and mixes with equimolar concentrations of gRNA and trRNA (150 pmol) were incubated for 5 min at 95 °C. The duplexes were incubated for 15 min at 37 °C with TrueCut Cas9 Nuclease V2 (50 pmol; Thermo Fisher) to form ribonucleoprotein complexes. Purified CD4+ T cells from the small intestine were resuspended with the ribonucleoprotein complexes and the Electroporation Enhancer in P3 Primary Cell buffer (Lonza) just before electroporation with 4D Nucleofactor (Lonza, program DN100). Electroporated cells, stained with Atto550 fluorescent molecule, were recovered in RPMI medium supplemented with 10% FBS for 30 min before being injected i.v. in C57BL/6 mice. The following gRNA sequences were used: Klf6 5′-CACGAAACGGGCTACTTCT-3′ and control 5′-CGCGATAGCGGCGAATATATT-3′.
Quantitative PCR with reverse transcriptionmRNAs were isolated using an RNeasy mini kit (Qiagen) and reverse transcribed with an iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR with reverse transcription was performed using LightCycler 480 SYBR Green Master and different sets of primers on a LightCycler 480 Real-Time PCR System (Roche). Sample gene expression was normalized to the levels of Gadph and analyzed according to the ΔΔCt method. The following primer sequences were used: Tgfb1 forward 5′-CCCGAAGCGGACTACTATGC-3′ and reverse 5′-ATAGATGGCGTTGTTGCGGT-3′; Gadph forward 5′-GCATGGCCTTCCGTGTTC-3′ and reverse 5′-TGTCATCATACTTGGCAGGTTTCT-3′.
Enzyme-linked immunosorbent assay (ELISA)Tissue sections 1 cm in length of the different intestinal segments were placed into RPMI medium (Gibco) containing 2% FBS (Gibco) and incubated for 24 h at 37 °C with 5% CO2. Supernatants were collected and stored before analysis at −80 °C. IL-12p70, IL-23p19 and IL-22 were measured by ELISA according to the manufacturer’s instructions (Invitrogen, BMS616 (IL-12p70); Invitrogen, BMS6017 (IL-23p19); Biolegend, 436304 (IL-22)). Optical density was read using a TECAN Infinite m1000 microplate reader at 450 nm.
Mapping human and mouse KLF6 binding sites on TBX21Human and mouse (TBX21 and Tbx21, respectively) loci were screened for KLF6 binding sites, and their respective chromosomal locations (mm10 and hg38 assemblies, respectively) were used to obtain Tbx21 DNA sequences extended by 15,000 bp up- and downstream (GenomicFeatures package). Sequences were then scanned for TF motifs from the MotifDb annotated collection using the Biostrings package function ‘match PWM’.
KLF6 expression in donor TH17 cellsCD45+ cells from (1) cerebrospinal fluid from individuals with multiple sclerosis and healthy donors, (2) colonic biopsies from individuals with ulcerative colitis and healthy donors or (3) ileal biopsies from individuals with Crohn’s disease in inflamed and not inflamed regions were analyzed by scRNA-seq47,48,49. For multiple sclerosis, we used the data from all six individuals and six healthy donors48. For ulcerative colitis, we used data from three individuals and three healthy donors49. For Crohn’s disease, we used data from nine individuals containing inflamed and noninflamed regions of the ileum of a same patient47. For all samples, we first identified the CD4+ T cell population. Gene expression signatures for TH17 cells and Treg cells were then applied to determine the TH17 and Treg cell clusters (Supplementary Table 5) using R software with AddModuleScore UCell packages. KLF6 expression levels in TH17 cells were determined and normalized to those in Treg cells from the same donor.
StatisticsAll statistical analyses were performed using Prism v9.4.1 (GraphPad) except for permutational multivariate analysis of variance, for which Qiime2 2020.8 was used. Statistical relevance was evaluated using an unpaired t-test or Mann–Whitney test when appropriate. Data distribution was assumed to be normal (unless stated otherwise), but this was not formally tested. Mice were chosen randomly for the experiments. Differences were considered significant when P values were <0.05. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications62. No data points nor animals were excluded from the analysis. Experimental conditions were organized randomly. Data collection and analysis were performed blind to the conditions of the experiments for histology scores.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)