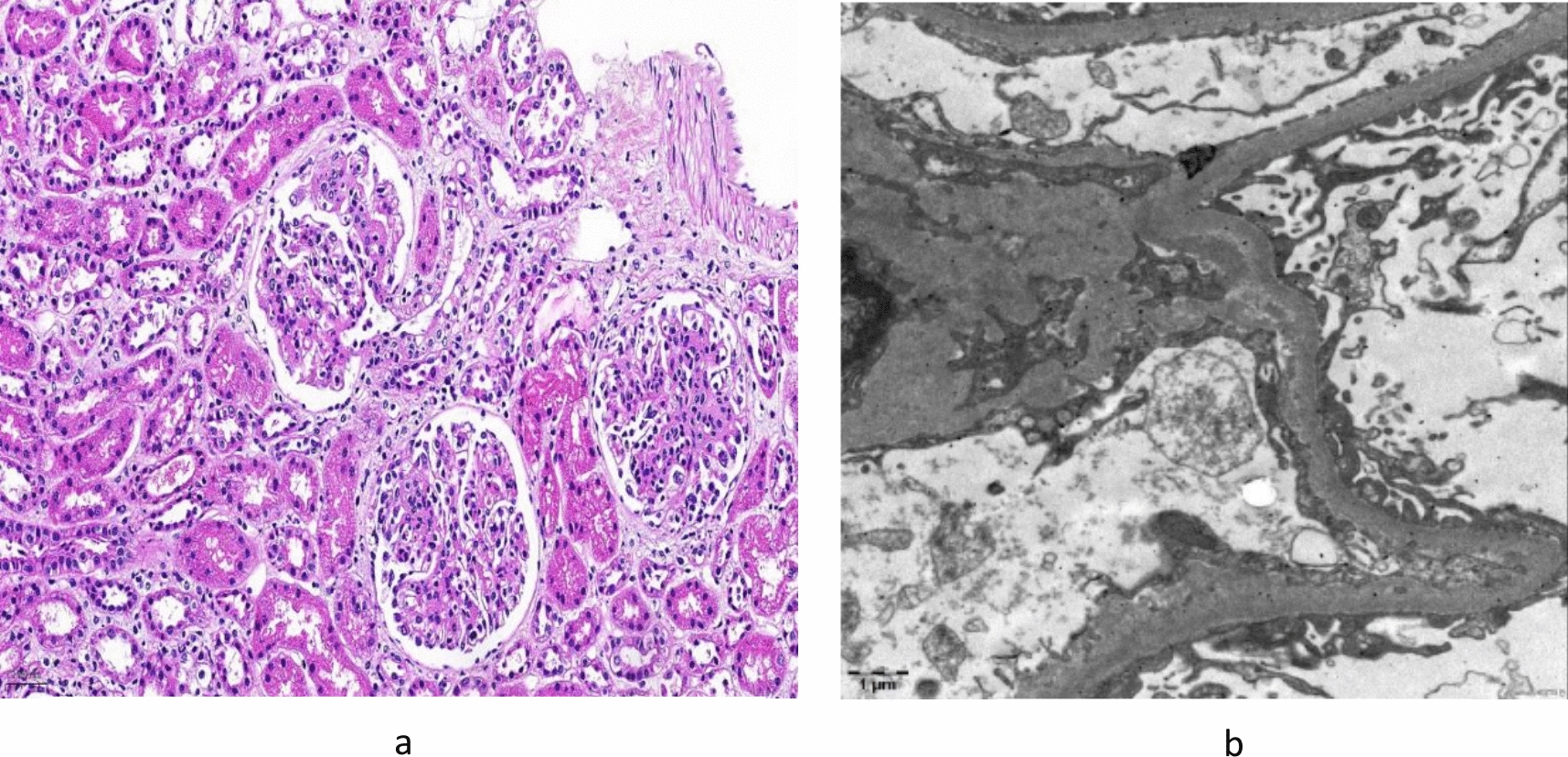
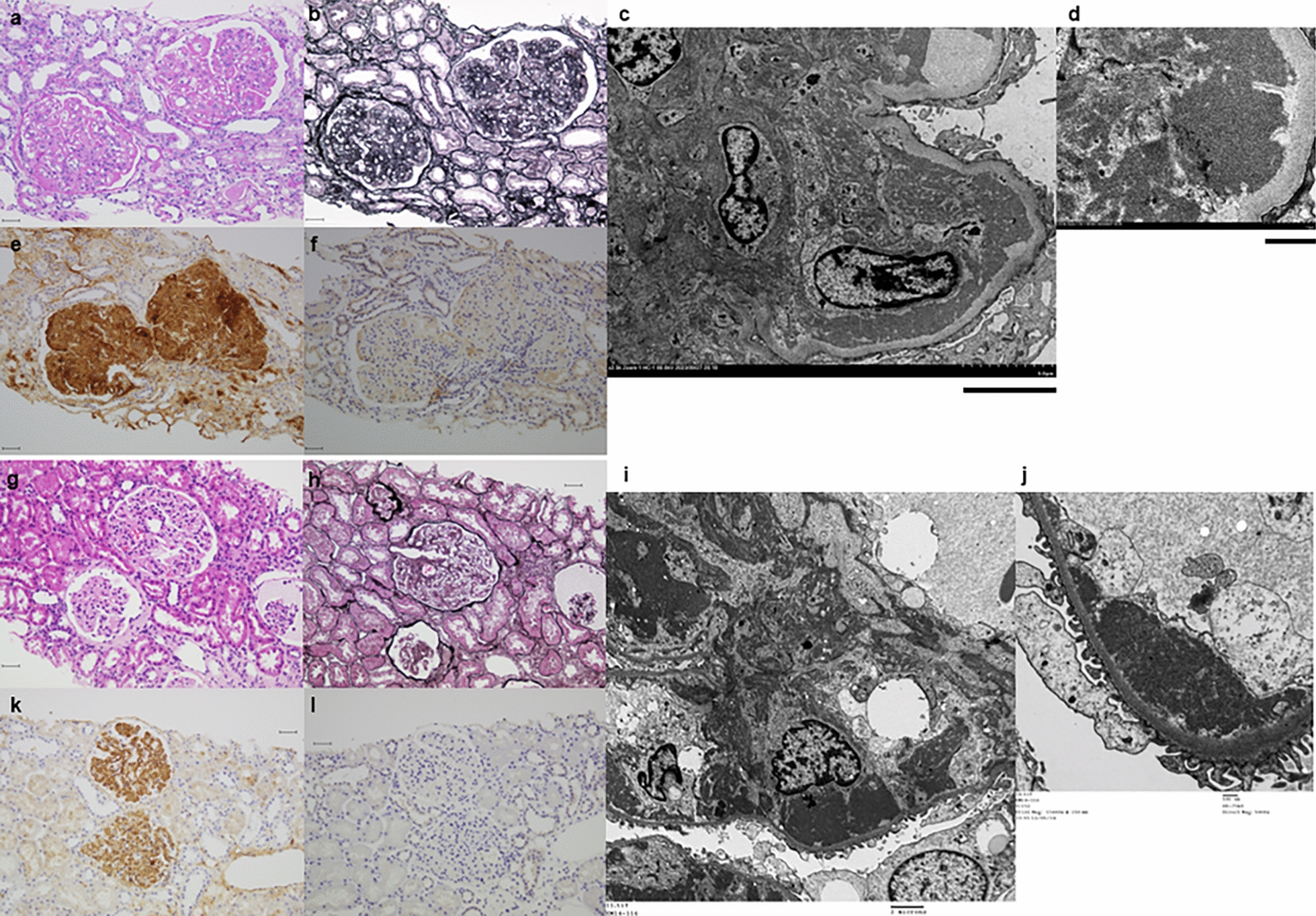
We identified a single family with a heterozygous gene variant of the PAX2 gene in exon 8 (c.959C > G). The phenotype of renal symptoms was presumably caused by PAX2 gene variants. In this case, oligonephronia with few glomeruli was observed on the open renal biopsy tissue. More than half of all glomeruli had a diameter > 200 μm, indicating glomerular hypertrophy. In oligonephronia, the pathogenesis of secondary FSGS is associated with glomerular hypertrophy owing to glomerular hyperfiltration [8]. The risk of oligomeganephronia owing to the PAX2 variant could not be ruled out in this case. The patient also had been a preterm baby at 33 weeks 2 days of gestation, and his birth weight was 1768 g. Nephron formation in humans begins at 9 weeks and ends at 34–36 weeks of gestation; however, there was no evidence of postnatal nephrogenesis. Perinatal abnormalities, preterm birth, and low birth weight are known risk factors for decreased nephron counts, and infants exposed to low nutrition during this period in utero reportedly have low birth weights and low estimated glomerular filtration rates as adults [9, 10]. The patient had an appropriate birth weight relative to weeks of gestation, and it was unlikely that the patient received poor nutrition before birth. Although it is possible that the preterm birth itself had something to do with the decrease in the number of nephrons, nephron formation is almost complete at 33 weeks of gestation, and it is unlikely that the extreme decrease in the number of glomeruli led to secondary FSGS. The low Apgar score at birth also suggests that renal ischemia owing to circulatory failure immediately after birth may have reduced the number of nephrons. Therefore, the possibility of secondary FSGS due to oligomeganephronia owing to PAX2 variants or perinatal abnormalities cannot be completely ruled out. However, since the patient had severe proteinuria and there was a family history of FSGS in the paternal side of the family, familial FSGS caused by PAX2 gene variants was considered.
Further, GBM changes, such as laminar variations, similar to AS, were observed on TEM. In the study of Ohtsubo et al., GBS Alport-like changes were observed in sisters with RCS associated with PAX2 gene variants in exon 2 (c.76dup, p.Val26Gly fsx27) [5]. The GBM comprises a basic skeleton of type IV collagen, with a combination of laminin, proteoglycans, and entactin are combined. Podocytes are combined by avβ3 integrin to type IV collagen α-chains that comprise the GBM. Podocytes produce and repair the GBM.
PAX2 is a transcription factor expressed in the Wolffian tube and ureteric buds in the embryonic kidney. Among the transcription factors, PAX2 and WT1 have important roles in podocyte maturation [11]. In this case, exon 8 with a heterozygous variant corresponded to the transactivation domain [12]. Therefore, we concluded that podocytes associated with PAX2 gene exon8 also may have caused abnormal production and repair of basement membranes, resulting in Alport-like changes. However, there have been no previous reports of exon 8 abnormalities in relation to significant changes in the GBM as evaluated using an electron microscope. Further studies are required to clarify GBM changes caused by the PAX2 variant.
In the current case, LVSEM of the GBM revealed coarse meshwork changes. In the previous reports, the normal GBM exhibited thin and smooth on LVSEM (Fig. 3b) [6]. Moreover, in AS, the basket weave structure of the GBMs can be observed on LVSEM [7]. GBM changes caused by PAX2 variants may be similar to those in AS on TEM. However, they may present with coarser changes compared with the reticular structure in AS on LVSEM, which provides a three-dimensional structural view. LVSEM can visualize pathophysiological differences. AS is a structural abnormality caused by variants in type IV collagen in the GBM. Meanwhile, the PAX2 variant is an abnormality of the GBM caused by podocyte irregularities. Hence, more cases should be collected and assessed in the future.
In conclusion, familial FSGS with Alport-like GBM changes caused by the PAX2 variant was diagnosed in an 8-year-old boy with proteinuria and decreased renal function.






Comments (0)