
Remember me
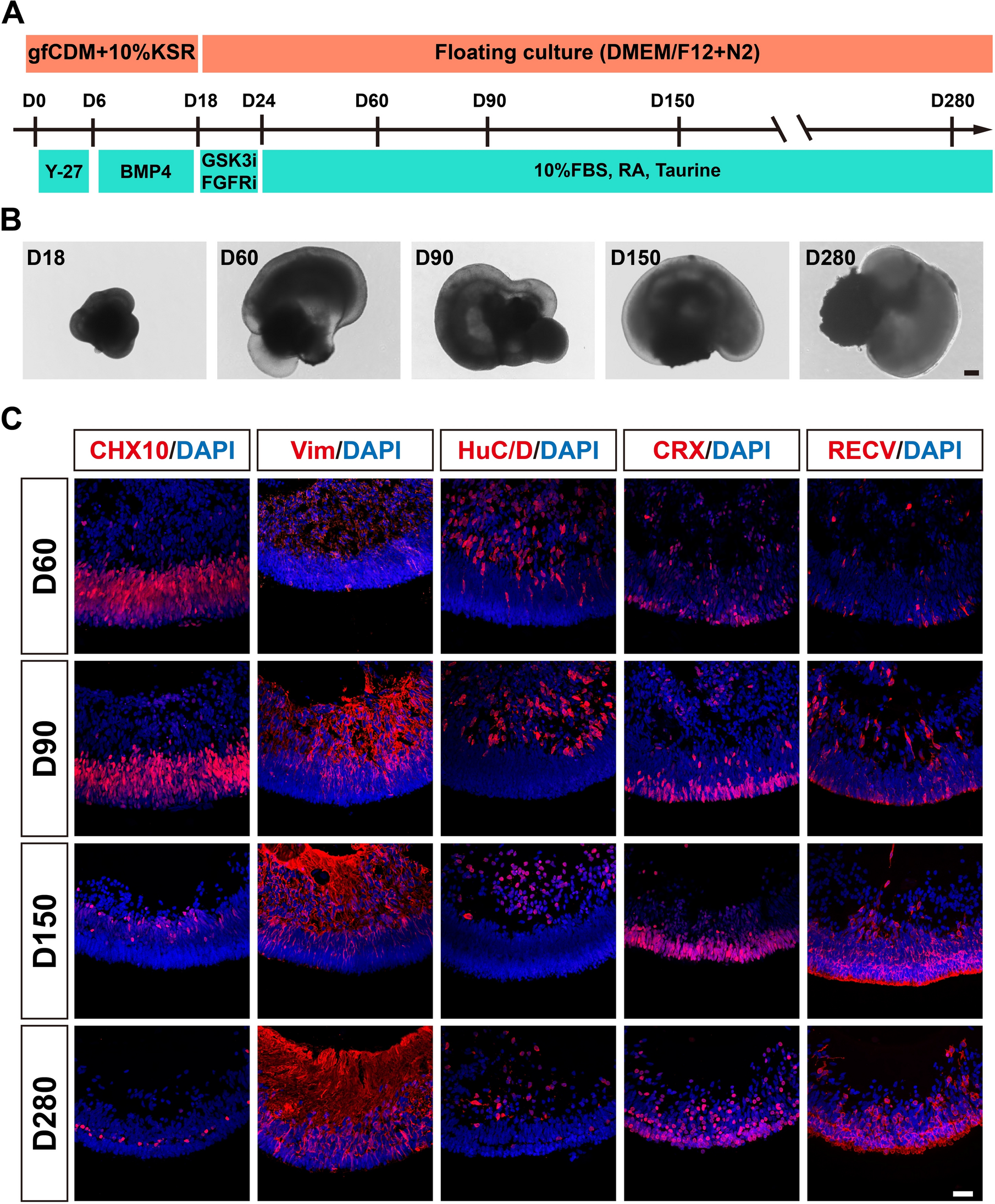







The study design was illustrated in Fig. 1. Of 25 included patients, 14 (56%) were well response to UCDA. 11 (44%) PBC patients underwent a poor response to UCDA after 12-months treatments. Supplementary Table 1 shows the baseline characteristics for the total population and for PBC patients with and without response. The mean age was 53.42 ± 10.17 years, and most patients were female (80%). Significant differences at baseline were found for TBIL (13.06 ± 3.99 vs 41.87 ± 27.01, P = 0.005), ALT (29.36 ± 16.57 vs 67.91 ± 34.79, P = 0.005), AST (39.29 ± 24.64 vs 85.82 ± 45.42, P = 0.008), ALP (140.79 ± 71.62 vs 295.17 ± 170.04, P = 0.015), TBA (12.84 ± 12.32 vs 51.83 ± 47.13, P = 0.022). The mean Sex, Age, GGT and ALB were not significantly different between groups.
Fig. 1
Diagram of the study design
Taxonomic changes in gut microbiome in PBC patients with poor responseFirst, the microbial diversity of the two groups was analyzed. For alpha diversity, significant differences were found in the Chao index and Shannon index of bacteria between the PBC patients with and without response at the species level, but no significant differences were found in the Simpson index (Fig. 2A, Supplementary Fig. 2A). At the genus level, Chao index was significant different between the two groups, while Simpson index and Shannon index showed no significant differences (Fig. 2B, Supplementary Fig. 2B). To further identify the overall microbial features of the two groups, beta diversity comparison and PCoA based on Bray–Curtis distance were performed. The results showed that the overall bacterial community structure of PBC with poor response was significantly different from PBC with response at the species level (Anosim test, P = 0.0086) and genus level (Anosim test, P = 0.0175) (Fig. 2C–E, Supplementary Fig. 2C). These results suggested that the taxonomic diversity with respect to its richness and evenness were not significant different between the two groups.
Fig. 2
Fecal microbiome variations in PBC with poor response (the response n = 14 versus the none response n = 11 n represent biological replicates). A Alpha diversity comparison of two groups at the species level; B Alpha diversity comparison of two groups at the species level; C Beta diversity comparison of two groups; D The PCoA and NMDS overall bacterial community structure of the two groups at the genus level; E The PCoA and NMDS overall bacterial community structure of the two groups at the species level. *P < 0.05
Next, the microbial composition at different taxonomic levels was analyzed. The microbial composition at the species and genus level was shown in Fig. 3A and B. 30 taxa showed the most significantly differentially altered at the genus level and 30 taxa showed the most significantly differentially altered at the species level in the poor response group (Fig. 3C and D, Supplementary Tables 2, 3). Microbiota belongs to Gemmiger, Prevotella, Ruminococcus_B and Clostridium genera showed significantly different between the two groups (Fig. 3E), and Gemmiger_qucibialis, Bariatricus_comes, Faecalibacterium_prausnitzii, Blautia_A_obeum, CAG-41_sp900066215 and Prevotella_sp900557255 were significantly decreased, and Ruminococcus_B_gnavus was significantly increased in the poor response group at the species level (Fig. 3F). The differential microbiota showed in the Fig. 3F were the differential microbiota for the further research.
Fig. 3
Gut microbiota signatures in PBC with poor response (the response n = 14 versus the none response n = 11, n represent biological replicates). A The composition of microbiota in the two group at the genus level; B The composition of microbiota in the two group at the species level; C Boxplots show the relative abundance of taxa exclusively altered in PBC with poor response at the genus level; D Boxplots show the relative abundance of taxa exclusively altered in PBC with poor response at the species level; E Stampplot show the relative abundance of taxa exclusively altered in PBC with poor response at the genus level; F Stampplot show the relative abundance of taxa exclusively altered in PBC with poor response at the species level
Changes in microbial function in PBC patients with poor responseFirst, we assessed the microbial function annotated by the KO database. According to the KO database, the overall microbiota transcript function of the poor response group was not significantly different from the response group (Anosim test, P = 0.063) (Fig. 4A). There were 9 functions, including enolase [EC:4.2.1.11], elongation factor Tu, molecular chaperone DnaK, glyceraldehyde 3-phosphate dehydrogenase (phosphorylating) [EC:1.2.1.12], ATP-dependent Clp protease ATP-binding subunit ClpL, formate C-acetyltransferase [EC:2.3.1.54], chaperonin GroEL [EC:5.6.1.7], glucose-1-phosphate adenylyltransferase [EC:2.7.7.27] andelongation factor G, showed mainly absolute abundance in both the two group (Fig. 4B, Table 1). Among these 9 functions, elongation factor Tu and elongation factor G were significantly increased in the poor response group (Fig. 4C, Supplementary Table 4, Supplementary Fig. 3A). To further elucidate the association between the microbiota and microbiota function, spearman analysis was carried out. Gemmiger_qucibialis, CAG_41_sp900066215 and Prevotella showed the significant negatively relationship with both elongation factor Tu and elongation factor G (Fig. 4D, Supplementary Table 5). These results suggested the effect of bacterial function to the host.
Fig. 4
Microbiota function in PBC with poor response (the response n = 14 versus the none response n = 11, n represent biological replicates). A The overall microbiota transcript function of the two groups based on the KO database; B The overall microbiota function community structure of the two groups based on the KO database; C The significantly different microbiota function between the group based on the KO database; D Spearman analysis showed the relationship between differential microbiota and microbiota function based on the KO database; E The overall microbiota transcript function of the two groups based on the swissprot database; F The overall microbiota function community structure of the two groups based on the swissprot database; G The significantly different microbiota function between the group based on the swissprot database; H. Spearman analysis showed the relationship between differential microbiota and microbiota function based on the swissprot database. * < 0.05
Table 1 The mainly absolute abundance in both the two group. based on the KEGG database (https://www.kegg.jp/kegg/kegg2.html)Then, we assessed the microbial function annotated by the SWISS-PROT database. According to the SWISS-PROT database, the overall microbiota transcript function of the poor response was also not significantly different from the response group (Anosim test, P = 0.0682) (Fig. 4E). There were 10 proteins, including RNA replication protein, Movement protein, Enolase, Capsid protein, Replicase large subunit, Probable ATP-dependent Clp protease ATP-binding subunit, Capsid protein, Movement protein and Pyruvate, phosphate dikinase, showed mainly absolute abundance in both the two group (Fig. 4F, Table 2). Among these 10 proteins, RNA replication protein was significantly decreased, while the Replicase large subunit, Capsid protein and Movement protein were significantly increased in the poor response group (Fig. 4G, Supplementary Table 6, Supplementary Fig. 3B). Spearman analysis was carried out to elucidate the association between the microbiota and microbiota function. Bariatricus_comes showed the significantly positively relationship with Replicase large subunit, Capsid protein and Movement protein, while significantly negatively related to RNA replication protein. Ruminococcus_B showed the significant negatively relationship with Replicase large subunit, Capsid protein and Movement protein. RNA replication protein showed the significant negatively relationship with Gemmiger_qucibialis and CAG_41_sp900066215 (Fig. 4H, Supplementary Table 7). These results suggested the bacterial function effected by the host gut environment.
Table 2 The mainly absolute abundance in both the two group. based on the swissprot database (https://www.uniprot.org/)Microbiota derived bile salt hydrolase (BSH) was identified as one of the most priority enzyme in BAs metabolism [10]. So, we also assessed the level of the BSH in the two groups annotated by the KO database. While there was no significant difference in the abundance of BSH between the two group (Supplementary Fig. 4A). The different phylotypes of microbiota derived BSH was retrieved in the UniProt database (https://www.uniprot.org/). BSH derived from 9 kinds of Lactobacillus genera, 4 kind of Bifidobacterium genera and Blautia_obeum were detected (Supplementary Table 8). The Blautia_obeum, which was the significantly decreased species in the poor response group mentioned above, producing lower level of BSH (uniport ID A0A174NYZ7) (P = 0.048) in the poor response group. In addition, The Lactobacillus_plantarum (uniport ID M1R991) (P = 0.034) derived BSH was significant lower, while the gene of Lactobacillus_salivarius (uniport ID C7AQY2) (P = 0.045) and Bifidobacterium_longum (uniport ID Q9KK62) (P = 0.009) derived BSH were significant higher in the poor response group (Supplementary Fig. 4B, Supplementary Table 8). These results suggested that the level of BSH may not affect the prognosis the PBC.
Taken together, these results indicate notable changes in the microbial function in PBC patients with poor response, which may contribute to disease development.
Associations of fecal metabolites in PBC patients with poor responseTo further elucidate the alteration of bacterial metabolism, we performed untargeted metabolomics using the fecal samples. Compared with the response group, there was significant overall distribution of metabolite in the poor response group according to the orthogonal partial least squares discriminant analysis (OPLS-DA) (Supplementary Fig. 5A). In order to judge the quality of the model without fitting risk, 200 response permutation tests were performed on the OPLS-DA model (Supplementary Fig. 5B). Based on the differential metabolites screening criteria: (1) VIP ≥ 1; (2) Fold Change ≥ 1.2 or ≤ 0.83; (3) p-value < 0.05. There was 1223 up regulated metabolites and 1281 down regulated metabolites in the poor response group compared to the response group. By screening BMDB, HMDB, KEGG, lipidmaps, Masslist and mzCloud database, we screened out 182 metabolites that were critical for host physiological progress. The results showed significant decreased pathway in Nucleotide metabolism, Bile secretion and Histidine metabolism between the two groups. (Fig. 5A, B, Supplementary Table 9). These results suggested that the multiple differential metabolites between the two groups.
Fig. 5
The fecal metabolite in PBC with poor response (the response n = 14 versus the none response n = 11, n represent biological replicates). A The number of differential metabolites in the poor response group compared to the response group; B The bubble chart of metabolites pathway between the two group. The X-axis Rich Factor is the number of differential metabolites annotated in this Pathway divided by all identified metabolites annotated in this Pathway. The higher the value is, the higher the ratio of differential metabolites annotated in this Pathway is. The dot size represents the number of differential metabolites annotated in this Pathway; C BAs metabolism related pathway enrichment analysis network plot. The circles represent metabolic pathway, and the triangles represent metabolites, and rhombus represent the class of the metabolites. Red indicates up-regulation and yellow indicates down-regulation; D The rank sum test showed the primary BAs and secondary BAs in the two groups; E The rank sum test showed the BAs pool in the two groups; F Immune related pathway enrichment analysis network plot. The circles represent metabolic pathway, and the triangles represent metabolites, and rhombus represent the class of the metabolites. Yellow indicates down-regulation
Dysbiosis of BAs metabolism is one of the main features of PBC. Then, we assessed the differential metabolites related to BAs metabolism. There were 17 differential metabolites relating to the altered BAs metabolism in PBC with poor response, including L-Cysteine, Pyruvic acid, [5-L-Glutamyl-taurine 5-Glutamyl-taurine Glutaurine], Leukotriene B4, Thromboxane B2, Oxoglutaric acid, Choline, D-( +)-Glucose, Glycochenodeoxycholic acid 3-glucuronide, Bilirubin, Chenodeoxycholic acid glycine conjugate, Cholic acid, Taurine, Deoxycholic acid, Lithocholic acid and Estrone sulfate. These metabolites belong to five class, including Organic acids and derivatives, Lipids and lipid-like molecules, Organic nitrogen compounds, Carbohydrates and Organoheterocyclic compounds. These metabolites effect four BAs related pathway, including map00430 (Taurine and hypotaurine metabolism), map04976 (Bile secretion), map00120 (Primary bile acid biosynthesis) and map00121 (Secondary bile acid biosynthesis). (Fig. 5C, Supplementary Figs. 7–10, Supplementary Table 9) Base on the rank sum test, we found the higher level of primary BAs (P = 0.025, Mann–Whitney U 36) and lower level of secondary BAs (P = 0, Mann–Whitney U 2) in the poor response group compared to the response group (Fig. 5D and Supplementary Fig. 6). In the BAs pool, the significant lower level of Cholate (P = 0.001, Mann–Whitney U 21) and Taurocholic acid (P = 0.018, Mann–Whitney U 57), as well as significant higher level of Lithocholic acid (P = 0, Mann–Whitney U 2) were detected in the response group (Fig. 5E and Supplementary Fig. 6). Dysbiosis of immune system is also one of the main features of PBC. Then, we assessed the differential metabolites related to abnormal immune response. The all-trans-Retinoic acid is decreased in the poor response group. The all-trans-Retinoic acid belongs to the Lipids and lipid-like molecules class and related to two pathways related to immune progression, including map04659 (Th17 cell differentiation) and map04672 (Intestinal immune network for IgA production) (Fig. 5F and Supplementary Figs. 11–12 and Supplementary Table 9). These results showed the abnormal metabolites related to BAs metabolism and immune response in PBC with poor response.
Next, we performed correlation analysis to investigate the associations between differentially abundant bacteria and metabolites. We found that bacteria enriched in the response group had strongly positive correlation with the response group-enriched metabolites but were negatively correlated with metabolites enriched in the poor response group (Fig. 6A and Supplementary Table 10). Notably, we found that some less abundant secondary BAs (e.g. Lithocholic acid) and more abundance of primary BAs (e.g. Taurocholic acid) were positively correlated with differential bacteria in the poor response group mentioned above (Fig. 6B and Supplementary Table 11).
Fig. 6
The spearman analysis showed the relationship between microbiota and metabolites (A), microbiota and BAs pools (B). * < 0.05; n = 25, n represent biological replicates
Taken together, these results indicate the notable changes in the metabolites, the effects of altered metabolites on physiological process, and the relationship between differential expressed microbiota and metabolites in PBC with poor response, which may contribute to disease development.
Associations of the altered microbes and metabolites with clinical parametersThen the associations of clinical parameters and differentially abundant bacteria or metabolites were carried out. The differential expressed bacteria showed significant correlations with ALT, TBIL and total BAs, which were in accordance with previous study. For instance, Gemmiger_qucibialis was negatively correlated with ALT, TBIL and total BAs (Fig. 7A and Supplementary Table 12). 10 metabolites, including L-Cysteine, 5-L-Glutamyl-taurine, all-trans-Retinoic acid, Estrone sulfate, Taurine, D-( +)-Glucose, Glycochenodeoxycholic acid 3-glucuronide, Leukotriene B4 and Thromboxane B2, were positively associated with ALT, AST, TBIL, GGT, ALP and TBA. Pyruvic acid was negatively ALT, GGT and ALP (Fig. 7B and Supplementary Table 13). For instance, L-Cysteine was positively correlated with ALT, AST, TBIL, GGT, ALP and TBA. These findings suggest the effect of gut microbiota and bacterial metabolites in liver function parameters in PBC with poor prognosis, which may contribute to disease development.
Fig. 7
The spearman analysis showed the relationship between clinic indictors and microbiota (A), clinic indictors and metabolites (B). * < 0.05; n = 25, n represent biological replicates
Classifier discriminating PBC with poor response from PBC with responseThe value of using the differential microbiota and metabolites as biomarkers for the differential diagnosis of PBC with poor response was assessed. Through the receiver operating characteristic (ROC) curve (AUC > 0.85), we identified 1 bacterial genera, 2 bacterial species and 9 metabolites that could distinguish PBC patients with poor response from patients with response (Fig. 8).
Fig. 8
Disease classification based on the ROC plot. X-axis represents 1-specificity, y-axis represent sensitivity. The area under the curve is the AUC value. A higher AUC value indicates a more suitable metabolite as a biomarker
Comments (0)