
Remember me
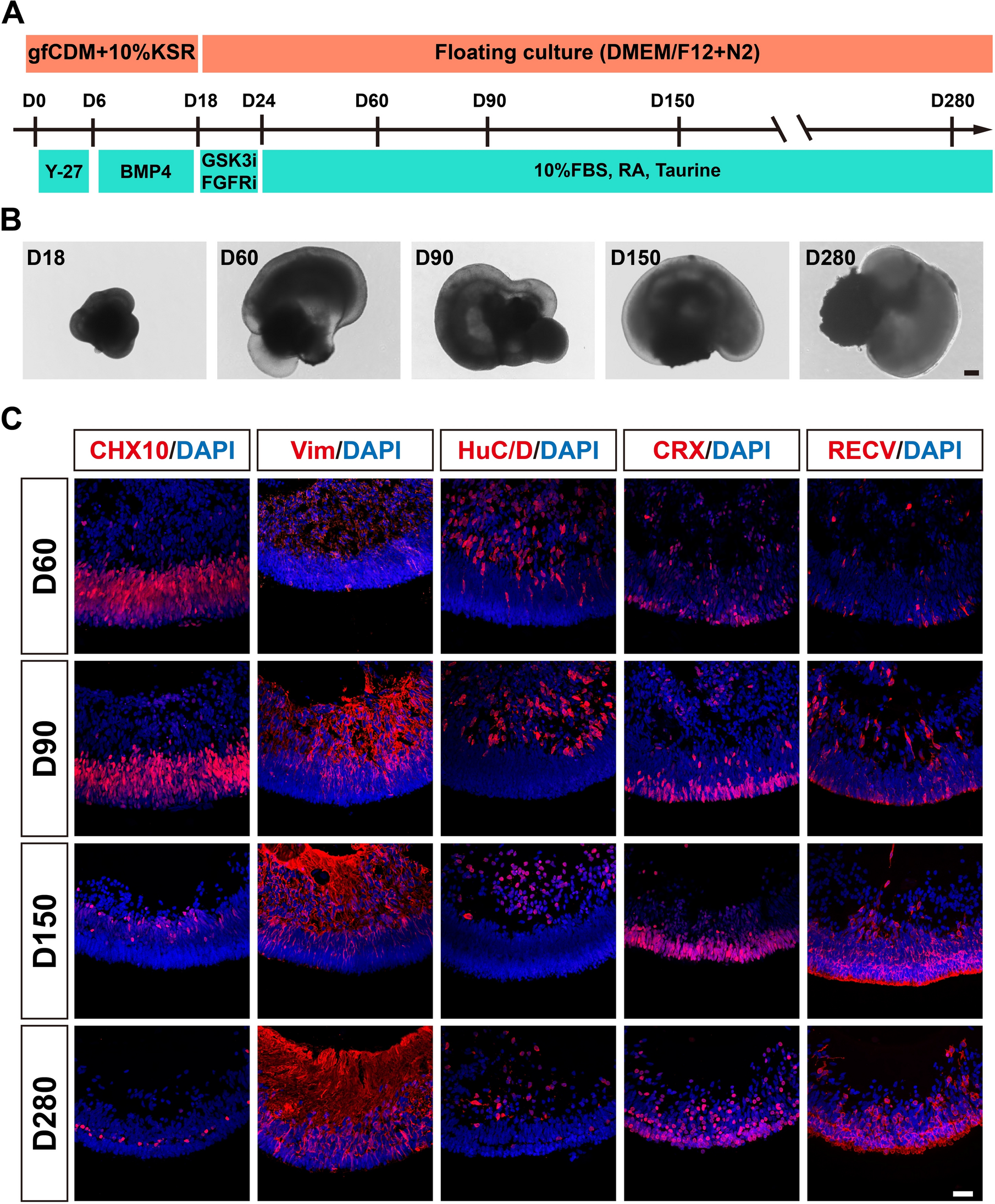







We used microarray technology to study the genome-wide gene expression profiles in differentiated NPCs, obtained from embryo telencephalons (E13; Fig. 1A). Neurospheres were obtained from WT and APPswe/PS1dE9 mice. The transgenic neurospheres were divided in three groups: untreated (APP_WT), treated with BK (BK_APP), and treated with HOE-140 (HOE_APP). The different neurosphere models presented a total of 3,639; 978 and 835 (p < 0.05) transcripts with differences in gene expression between neurospheres WT and APP_WT, HOE_APP and APP_WT and BK_APP and APP_WT, respectively.
Our results showed a specific enrichment in gene expression of APP_WT neurospheres, especially for genes related to immune responses (first column of Fig. 1B). This result corroborates the data published in our previous paper [7]. Controls used for this analysis are the same ones that allowed us to obtain consistent data in our previous work, and were reanalyzed for the data reported here.
In our current study, we found that BK treatment (BK_APP neurospheres) increased the expression of genes related to immune responses (Fig. 1B, second column). Conversely, expression levels of these genes was decreased in HOE_APP neurospheres (Fig. 1B, third column).
Fig. 1
Workflow of neurosphere preparation and differentiation, and heatmap of gene relative expression. A Schematic representation of fundamental steps for performing neurosphere assays obtained from the telencephalon of mouse embryos (E13). B Altered gene expression profiles comparing APP_WT with WT neurospheres; BK_APP with APP_WT neurospheres and HOE_APP with APP_WT neurospheres. Heatmaps showing the expression ratios (log2) of genes related to the immune response of four independent replicates (R1-R4) of the comparison described above. Differentially expressed transcripts were identified using SAM and rank products (p ≤ 0.05)
Expression changes in neurospheres treated with BK and HOE-140To understand the effects of BK and HOE-140 treatments on differentiated AD neurospheres, we determined the genes, which were significantly enriched in pathways related to immune responses. Comparison between the expression in APP_WT and HOE_APP genes demonstrated 18 genes, which were underexpressed: bone marrow stromal antigen 2, complement C4-B, C-C motif chemokine 5, CX3C chemokine receptor 1, C-X-C motif chemokine 10, antiviral innate immune response receptor RIG-I, guanylate-binding protein 5, guanylate-binding protein 6, psychosine receptor, interferon-induced helicase C domain-containing protein 1, interferon-induced protein with tetratricopeptide repeats 1, interferon-induced protein with tetratricopeptide repeats 3, ubiquitin-like protein ISG15, lymphocyte antigen 86, tyrosine-protein phosphatase non-receptor type 6, S-adenosylmethionine-dependent nucleotide dehydratase RSAD2, Toll-like receptor 2 and zinc finger CCCH-type antiviral protein 1 (BST2, C4B, CCL5, CX3CR1, CXCL10, DDX58, GBP5, GBP6, GPR65, IFIH1, IFIT1, IFIT3, ISG15, LY86, PTPN6, RSAD2, TLR2 and ZC3HAV1, respectively) in neurospheres treated with HOE-140 (Fig. 2A). Twenty-four genes were overexpressed in BK_APP compared to APP_WT: B-cell lymphoma 3 protein, B-cell linker protein, complement C1q subcomponent subunit B, caspase-4, C-C motif chemokine 2, C-C motif chemokine 3, C-C motif chemokine 4, monocyte differentiation antigen CD14, CX3C chemokine receptor 1, C-X-C motif chemokine 5, adhesion G protein-coupled receptor E1, fatty acid synthase, guanylate-binding protein 5, GTP cyclohydrolase 1, interferon-induced protein with tetratricopeptide repeats 1, immunoglobulin superfamily member 6, ubiquitin-like protein ISG15, leukocyte immunoglobulin-like receptor subfamily B member 4, Toll-like receptor 2, tumor necrosis factor alpha, triggering receptor expressed on myeloid cells 2, TYRO protein tyrosine kinase-binding protein, proto-oncogene vav and vascular cell adhesion protein 1 (BCL3, BLNK, C1QB, CASP4, CCL2, CCL3, CCL4, CD14, CX3CR1, CXCL5, EMR1, FAS, GBP5, GCH1, IFIT1, IGSF6, ISG15, LILRB4, TLR2, TNF-alpha, TREM2, TYROBP, VAV1 and VCAM1, respectively) in BK-treated neurospheres (Fig. 2B).
Among the 37 genes modulated by the treatments, five showed opposite behaviors, being overexpressed in conditions of BK stimulation and underexpressed when the B2 receptor had been blocked with HOE-140: CX3CR1, GBP5, IFIT1, ISG15, and TLR2.
Fig. 2
Gene expression changes in transgenic neurospheres treated with BK and HOE-140. A Heatmap with emphasis on genes related to immune responses, which were underexpressed in HOE_APP neurospheres. B Heatmap with emphasis on genes related to immune responses, which were overexpressed in BK_APP neurospheres
Quantitative expression analysis of genes of differentiated APPswe/PS1dE9 neurospheresThe expression of eight important genes linked to inflammatory responses and cell-to-cell communication in the CNS was investigated by RT-real time PCR. In a previous study of our group [7], the expression of these genes was significantly increased in APP/PS1neurospheres, and it was now our interest to evaluate the expression pattern of these genes in BK /HOE-140 treated neurospheres.
The results (Fig. 3) revealed a significant decrease (p < 0.05) in C3, CCL12, and CCL5 expression in HOE_APP neurospheres compared to APP_WT neurospheres. Expression of TLR2 and CCL12 was significantly increased (p < 0.05) in BK_APP neurospheres compared to APP_WT neurospheres. There were no significant differences in the expression of CCL3, CX3CR1, TNF, and Iba-1 genes between treated and untreated neurospheres.
Fig. 3
mRNA expression levels of 8 inflammatory genes in neurospheres. The experiments were performed by RT-real time PCR using at least four independent replicates. GAPDH gene expression was used to normalize expression levels (Paired t-test, two-tailed; ***p ˂0.05)
The presence of microglia in neurospheres is expected since these cells are known to migrate to the brain around the ninth day of embryonic development. Ginhoux and colleagues (2013) described that, with the establishment of the circulatory system, which occurs from E8.5 to E10, yolk-sac derived primitive macrophages would spread into the embryo through the blood, migrating to various tissues, including the brain, where they proliferate and colonize [23]. Since NPCs were isolated from embryo telencephalon (day 13; E13) of wild-type (WT) or APPswe/PSldE9 C57BL/6 mice embryos, macrophages already have migrated to the brain, where they differentiate into microglia. The RNA-seq analysis further corroborated the infiltration hypothesis. Microglial markers, including Aif1, Fcrls, Hexb, and Tmem119, were upregulated in their expression levels in APP/PS1 compared to WT neurospheres [7]. The Aif1 gene, that encodes the Iba protein, is also identified in the BK_APP. Expression of this microglial marker had a tendency of upregulation following BK treatment (Fig. 3). Additionally, samples from AD-differentiated neurospheres exhibited a higher frequency of CD11b-positive cells compared to those from WT animals, indicating the presence of activated microglia in this AD model (Fig. S1).
Reactome enrichmentThe human orthologous genes assigned to mouse genes detected in the microarrays and their respective FCs were used to analyze enriched pathways from Reactome and BPs and CCs from GO (Figs. 4 and 5). In GO, APP_WT displayed enrichment of 24 CCs and 135 BPs terms, HOE_APP genes were enriched in 26 BPs and BK_APP genes were enriched in 6 CCs and 57 BPs terms, respectively. No BP was found in common among the three groups (Fig. 4D). A similar analysis was performed for the enrichment of pathways in Reactome and CC in GO (Fig. 5). Only genes present in HOE_APP, and BK_APP groups generated significant enrichment of Reactome terms, with 3 and 16 enriched pathways, respectively.
Fig. 4
BPs by GO enrichment and their respective average FCs. A, B, and C, HOE_APP, APP_WT, and BK_APP genes, respectively. In the last two groups, only the 20 BPs with the biggest FCs are shown. In D the number of common processes between the groups is presented
Fig. 5
Enrichment of pathways by Reactome and CCs by GO and their respective average FCs. A and B pathways enriched in Reactome with all genes identified in BK_APP and HOE_APP, respectively; C and D, GO-enriched CCs with all genes identified in APP_WT and BK_APP, respectively
Correlation of gene expression between treatmentsBased on our hypothesis, it was possible to propose that HOE_WT, APP_WT, and BK_WT is the order that characterizes the increasing impairment of the immune system. To investigate this proposal, we verified which genes vary their expression following this order, with absolute Pearson’s correlation > 0.9 and p < 0.05, (Fig. 6). The calculation of FCs referring to WT is only possible among the genes identified in the three groups (HOE_APP, APP_WT, and BK_APP), therefore, this analysis was performed with 25 mouse genes and 17 human orthologous genes. Eight mouse genes and five human orthologs genes fit this restriction, all with positive correlations (Fig. 6A, and B, respectively).
Fig. 6
Proteins with an absolute Pearson’s correlation greater than 0.9 and a p-value lower than 0.05. A are arranged mouse proteins and in B their human orthologs
Construction of protein-protein interaction (PPI) networks and calculation of weighted system impact (wSI)The number of proteins and interactions in each PPI network are presented in (Fig. S2). The PC analysis in the HOE_APP, APP_WT, and BK_APP progression can also be done with the protein’s wSI, in order to highlight the role of treatments in the topological position of the proteins in the network (Fig. 7). Epithelial membrane protein 1, TNF receptor-associated factor 1, stimulator of interferon genes protein, synaptosomal-associated protein 29, syntaxin-4 and ubiquilin-1 (EMP1, TRAF1, STING1, SNAP29, STX4, and UBQLN1, respectively) in addition to having extreme PC values, are correlated with the immune system, with the first three being PC positive and the others being negative.
Fig. 7
Proteins and wSI, which have absolute Pearson’s correlation greater than 0.9. Lines in shades of red represent progression with negative PC and those in shades of green with positive PC (p-value < 0.05)
Comparison with the transcriptome of human AD patientsIn order to make a parallel between the findings determined through the murine neurosphere models and human patients, comparisons were made with a dataset of tissue from cases with Alzheimer’s available in the GEO database (GSE118553) [21]. The detected coding genes and their respective FCs between AD patients and normal controls were used for comparison with the transcriptomes of the neurosphere model and their treatments, in two ways: by FCs and wSI of the common genes between the groups. For these, PPI networks were constructed for each tissue and condition of patients with AD. Isolated sets were deleted and only the main set was used in this analysis.
The reorganization of the gene expression pattern generated by the treatments with HOE-140 and BK can be observed by the correlation of gene expression with the human profile (Fig. 8 and clusters in Table S2). Only 11 genes are shared between the neurosphere experimental groups and those obtained from the human dataset. The Pearson’s correlation values of these genes are moderately negative between the HOE_APP group and all regions of the human brain studied, except for the cerebellum, which has a low positive correlation. On the other hand, the APP_WT group mainly presents moderate positive correlations, and the BK_APP group has low positive correlations. This is an indication that treatment with HOE-140 generates a considerable change in the expression of these 11 transcripts compared to the expression in brain regions that AD compromises.
Fig. 8
The 11 genes were detected in the microarray analysis and in the human dataset. A correlation between FCs of genes. B heatmap with hierarchical clustering of FCs of the 11 genes commonly detected in the murine neurospheres and AD patients’ tissues
The wSI of each of the common proteins among all constructed networks was used to verify the correlation between the groups (Fig. 9 and Fig. S3). This analysis indicates whether the different groups compared have the “dominance” of the genes in the PPI network distributed in a similar way. BK_APP and HOE_APP have a lower to moderate correlation with the human dataset compared to APP_WT that has moderate to high correlations (Fig. 10A). This indicates that, in the topological context of the network, the APP_WT system favors proteins similarly to human disease. On the other hand, treatments with BK or HOE-140 generate a change in the model, mischaracterizing it, of its proximity to human tissues. The correlation of the APP_WT group is always higher for TC, FC, and EC tissues than with CB, which is consistent with the low or null impact of AD on CB. Among the three tissues affected by AD, the one with the highest correlation with APP_WT is EC, one of the first regions affected by the disease. For these same three tissues, the correlation of APP_WT with symptomatic patients is greater than with asymptomatic ones. Indicating that the model more closely represents patients with clinical diagnoses, advanced Braak stage, and neuropathological assessment at autopsy showed hallmark AD pathology.
Fig. 9
Correlation matrix between wSI of the genes. A: Correlation matrix of the 4211 genes contained in all groups. B: Correlation between wSI of the 59 genes belonging to the biological process innate immune response-activating signaling pathway. C: Correlation between wSI of the 82 genes belonging to the biological process immune response-activating cell surface receptor signaling pathway
The 4211 proteins common to all groups enrich, according to the Gene Ontology bank, two biological processes related to the immune response: innate immune response-activating signaling and immune response-activating cell surface receptor signaling pathways. The wSI of the genes that enrich these processes was used to construct a correlation matrix between the groups, (Fig. 9B and C). HOE_APP, APP_WT, and BK_APP, in this order, have a decreasing correlation with human tissues. That is, the treatment with HOE-140 alters the model of neurospheres bringing the topological position of the genes of HOE_APP closer to those of human tissues with AD. BK treatment decreases this correlation. This decreasing pattern and the correlations are significantly higher in the innate immune response-activating signaling pathway, indicating a targeted action of HOE-140 in this pathway. For this pathway and in HOE_APP, correlations are higher in symptomatic patients and especially in AD_EC, indicating that this treatment increases the dominance of genes affected by AD.
The genes present in the enriched processes related to the immune system were used to cluster the experimental groups and the tissues of patients with AD (Fig. 10 and clusters in Table S3). In both processes the APP_WT and BK_APP groups are part of the same group and HOE_APP is outside this group. In the innate immune response-activating signaling pathway (Fig. 10A), HOE_APP clusters with AsymAD_EC, AsymAD_TC, and AD_CB. Using the UMAP method, none of the asymptomatic groups of patients with AD were discriminable from healthy patients, (Fig. S4). Thus, the wSI of the genes of this biological process indicates that HOE_APP is closer to groups that are spared by AD (AD_CB) and to groups in which the effect of AD is not sufficient to differentiate patients from healthy controls. In this process, NFKBIA, CD40, MYD88, and IFH1 stand out for presenting significant differences between the groups (AsymAD_CB, AsymAD_FC, AD_FC, AD_TC, AD_EC) and (HOE_APP, AsymAD_EC, AsymAD_TC and AD_CB), p of 0.02, 0.0006, 0.04 and 0.004, respectively. In the immune response-activating cell surface receptor signaling pathway HOE_APP is isolated from all other groups, indicating a treatment-related change in the dominance of genes in this process.
Fig. 10
wSI clustered heatmap of proteins contained in GO BPs related to immune response. A wSI of the 59 genes belonging to the biological process innate immune response-activating signaling pathway. B wSI of the 82 genes belonging to the biological process immune response-activating cell surface receptor signaling pathway
Comments (0)