
Remember me






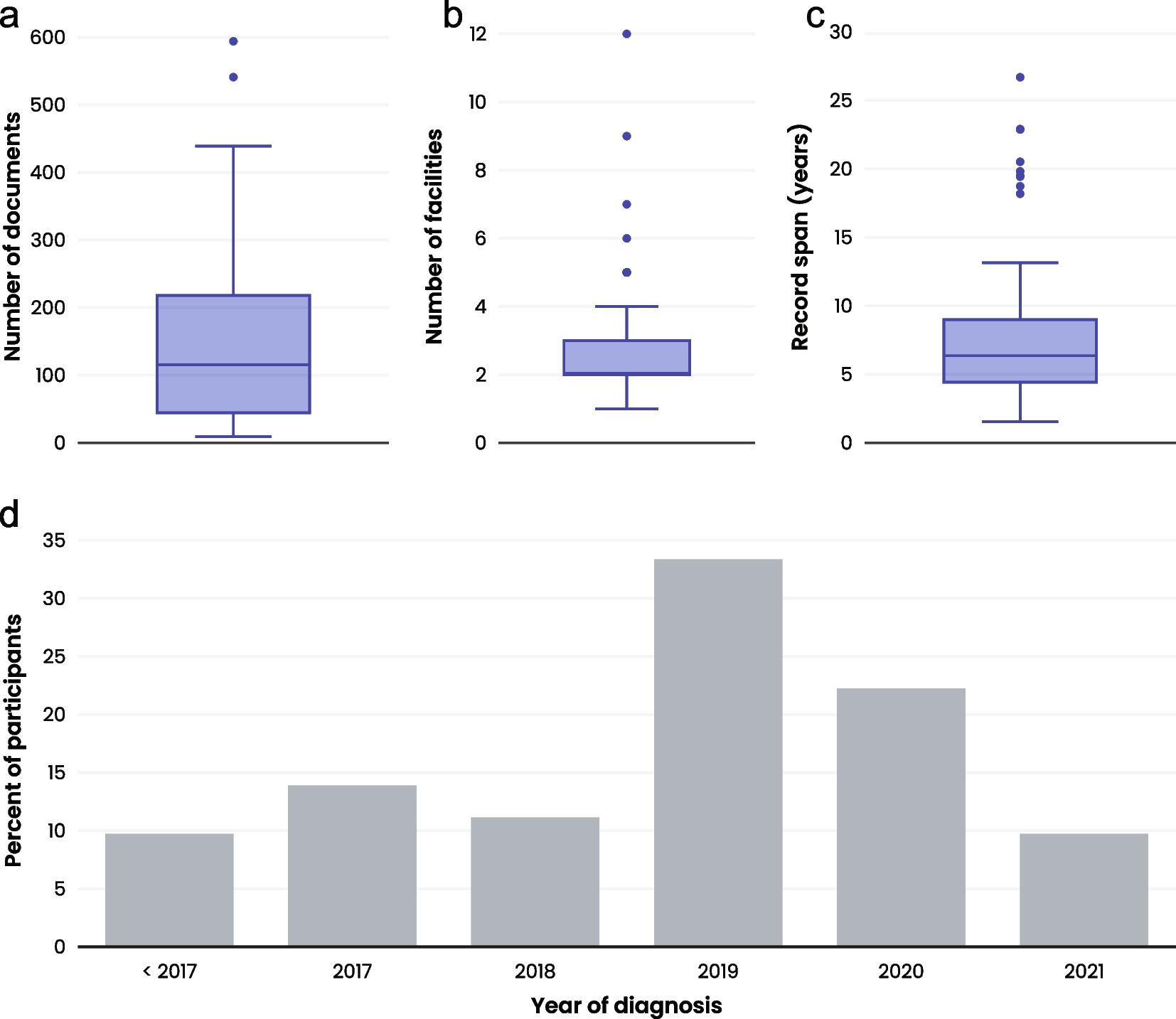

ES data from 80 patients (59 adults, 47 females) and GS from 20 patients (10 adults, 13 males) were analyzed. The age of the patients at the time of genetic testing ranged from 4 to 81 years old, with a mean age of 43 years. The original EGBP reports for these patients yielded the following results: negative in 54 patients, containing a variant of uncertain significance (VUS) in genes of interest in 41 patients, and reported as positive (containing one likely pathogenic or pathogenic variant in a gene associated with an AR phenotype) in 8 patients as demonstrated in Figure 1. The median between the issuance of the clinical report and the subsequent reanalysis of the sequencing data was 12 months, with an interquartile range (IQR) spanning from 7 to 30 months. The majority of patients were referred from the Nephrology division (n=44), followed by Rheumatology (n=29), Endocrinology (n=13), and Pulmonary and Critical Care Medicine (n=12). The most common reasons for testing were auto-inflammatory syndrome (n=30) and focal segmental glomerulosclerosis (FSGS) (n=26). The complete reason for referral can be found in Table 1. Patients of African ancestry (three individuals) were evaluated for the APOL1 (HGNC:618), G1 (NM_001136540:c.1024A>G, p.(Ser342Gly), and NM_001136540:c.1152T>G, p.(Ile384Met)) and G2 (NM_001136540:c.1160_1165delATAATT) polymorphic risk alleles due to the association with kidney disease within this population [15]. Demographic information can be found in Table 2 and Supplementary Table 2.
Fig. 1
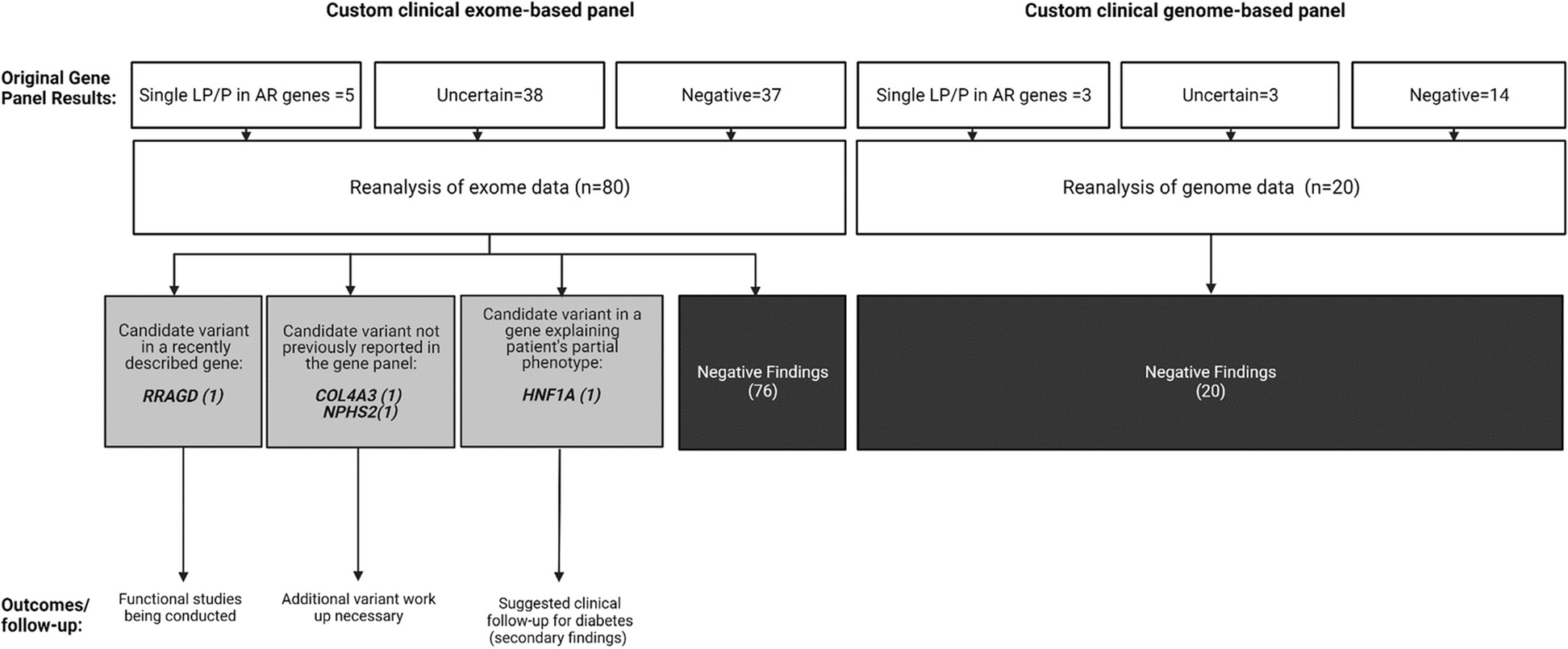
Results of the re-analyses of custom clinical exome and genome-based panels data of 100 patients with single-system diseases
Table 1 Phenotypes of the individuals included in the studyTable 2 Demographic informationUpon re-analysis of the exome/genome data, no additional findings were identified in 96 individuals. In the remaining four (4%), additional findings were discovered. In one case, a variant in the RRAGD (HGNC:19903) gene was found, which is associated with a phenotype reported in the literature after the release of the original report. In two cases, variants that were part of the original EGBP were not reported by the clinical laboratory. This included a COL4A3 (HGNC:2204) variant due to a discordant inheritance pattern and a variant in NPHS2 (HGNC:13394), which was omitted due to its high population prevalence. In a fourth case, a likely pathogenic variant in HNF1A (HGNC:11621) was identified, which might explain the patient's partial phenotype. The summary of the key learning points of each case can be found in Table 3. Additionally, periodic automated re-analysis during the specified period flagged variants in 26 cases; however, after further review, these variants were deemed not relevant for the proband's phenotypes since they were primarily single VUS in recessive genes or in genes associated with multisystem syndromes that were flagged by the softwares because those syndromes encompass HPO terms included in the referral reason (data not shown).
Table 3 Summary of the key learning points of the cases with findings after analysis of genomic raw dataCase vignettesCase 1 – conflicting inheritance patternA 62-year-old Caucasian female patient presents with a medical history characterized by focal segmental glomerulosclerosis (FSGS) lesion in a renal biopsy at the age of 57. Family history reveals two paternal uncles with kidney disease, attributed to congestive heart failure and diabetes, respectively. Initial symptoms manifested around age 56, marked by edema, with an albumin level of 2.8 g/dL (Reference Range, RR: 3.2 - 4.6 g/dL) and creatinine of 0.8 mg/dL (RR: 0.59 - 1.04 mg/dL). At 57, a 24-hour urine collection showed 9 g of protein (RR: <229 mg/24 h) and albumin levels of 1.8 g/dL (RR: 3.5 - 5.0 g/dL). The biopsy confirmed segmental glomerulosclerosis, with negative immunofluorescence for various markers except focal segmental immunoreactivity with fibrinogen (2+). Electron microscopy revealed extensive effacement of visceral epithelial cell foot processes. Commencing treatment with an angiotensin-converting enzyme inhibitor and prednisone, later switched to cyclosporin, the patient faced additional challenges such as mild hyperlipidemia, with triglyceride levels at 160 mg/dL (RR: <150 mg/dL). Baseline creatinine fluctuated between 2.2 to 2.6 mg/dL (RR: 0.59 - 1.04 mg/dL) since age 60. Despite interventions, renal function decline prompted enrollment in a clinical trial with obinutuzumab. Investigation for genetic causes of FSGS lesion with an EGBP was initiated at age 58 and yielded a negative result. Subsequent re-analysis of the sequencing data detected the NM_000091.4: c.3182 G>A; p.(Gly1061Asp), variant of uncertain significance (VUS) in COL4A3 (HGNC:2204), a gene associated with recessive and dominant forms of Alport syndrome (MIM 203780 and 104200). This glycine substitution is identified in 49 alleles out of 248,632, with no homozygotes in gnomAD. Notably, similar substitutions (p.(Gly1023Arg), p.(Gly1035Val), p.(Gly1038Ser)) have been described as pathogenic or likely pathogenic in the same exon. The variant was clinically confirmed by the laboratory after initial oversight due to conflicting inheritance patterns, and family segregation studies were recommended for a comprehensive understanding of the variant's role in the proband's phenotype, especially if other family members exhibit biopsy-proven FSGS.
Case 2- variant prevalent in the general populationA 43-year-old male with renal failure and FSGS lesion on the kidney biopsy. The diagnosis of FSGS was established at the age of 26 prompted by the discovery of proteinuria during an insurance screening, including urinalysis. Analysis of a 24-hour urine collection at that time revealed a protein loss of 7.7 g/24 h (reference range <229 mg/24 h). Further laboratory investigations disclosed hypercholesterolemia (total fasting cholesterol 320 mg/dL, desirable <200 mg/dL), hypertriglyceridemia (fasting triglycerides 576 mg/dL, reference range <150 mg/dL), and plasma albumin levels of 2.9 g/dL (reference range 3.4 to 5.4 g/dL). There was no familial history of similar symptoms. At the age of 38, an EGBP identified a pathogenic variant in exon 8 of NPHS2 (NM_014625.3: c.948delT; p. (Ala317LeufsTer31)) associated with autosomal recessive nephrotic syndrome type 2 (MIM 600995). This variant was deemed pathogenic by multiple clinical laboratories (ClinVar ID: 188990). The initial report did not mention a second hit in this gene, and the exome sequencing data showed no evidence of multi-exon deletion/duplication involving this gene. During quality control background testing for the EGBP, the clinical laboratory identified a likely duplication of the X chromosome, consistent with Klinefelter syndrome, a finding confirmed by karyotype analysis. This secondary discovery was considered causative for the patient's history of azoospermia and tall stature. Subsequent re-analysis of raw ES data uncovered a second variant in NPHS2 (HGNC:13394; NM_014625.4:c.686G>A; p.(Arg229Gln)), not previously reported by the clinical laboratory. Despite its prevalence in the general population (8,538 alleles out of 282,294 in gnomAD, including 186 homozygotes) and uncertain in silico predictions (REVEL = 0.58), this variant has been traditionally documented in the literature as disease-causing, depending on the variant observed on the other chromosome [16]. At the age of 42, the patient underwent a successful renal transplant from a living donor, experiencing an uneventful postoperative course with immediate kidney allograft function.
Case 3 – novel gene-disease associationA 46-year-old female of Ashkenazi Jewish descent with nephrolithiasis, hypomagnesemia, and hypokalemia. She has family history of the maternal grandmother experiencing nephrolithiasis, and her mother and two maternal uncles exhibiting electrolyte imbalances suggestive of Gitelman syndrome. Born via C-section at full term, her delivery was complicated by her mother's hypokalemia-induced cardiac arrest. At 8-10 months, she developed a urinary tract infection, with nephrolithiasis diagnosed at 15 months, necessitating a partial nephrectomy for stone removal. Throughout childhood, she frequently experienced urinary tract infections, responding well to sulfamethoxazole and trimethoprim therapy. In adulthood, recurrent severe pyelonephritis episodes ensued. Paresthesias developed, accompanied by intermittent hypokalemia, hypomagnesemia, and occasionally hypocalcemia. Treatment with magnesium and potassium replacement therapy was initiated. At 40, pancreatitis episodes exacerbated by pyelonephritis and sepsis led to a diabetes diagnosis, prompting a switch from metformin to insulin. A kidney ultrasound at 42 revealed medullary nephrocalcinosis with bilateral renal calculi, non-obstructive. Genetic testing at 43, conducted in November 2019 via EGBP, yielded negative results. However, a reanalysis of the ES data in January 2022 identified a VUS in RRAGD (Ras-related GTP binding D, HGNC:19903), a gene newly associated in November 2021 with hypomagnesemia, tubulopathy, and dilated cardiomyopathy. Variants in this gene have been described as causing electrolyte-losing tubulopathy and dilated cardiomyopathy due to the activation of mTOR signaling, suggesting a crucial role for Rag GTPase D in renal electrolyte regulation and cardiac function [17]. Patient’s variant is absent in population databases and has a high REVEL score of 0.67, predicting it to be deleterious. Despite normal echocardiogram results, further functional testing and family segregation studies are underway in collaboration with the original report authors due to the unique findings in this case [17].
Case 4 – secondary phenotypeA 44-year-old female with symptoms of hypokalemia and polyuria, with a notable family history of diabetes in her mother. The hypokalemia was initially identified at the age of 36 during an angina pectoris evaluation, prompted by an ECG revealing a prolonged QT interval. At that time, her potassium levels measured 2 mmol/L (reference range 3.6 - 5.2 mmol/L). Concurrently, she was diagnosed with polyuria, experiencing urine output ranging from 5 to 10 liters daily. Hypokalemic manifestations included intermittent neurological symptoms, reduced mentation, impaired concentration, dizziness, and headaches. Alongside hypokalemia, she presented with hypomagnesemia and chronic constipation, necessitating a rotating laxative regimen, possibly linked to her electrolyte imbalance. Her diabetes workup in 2021 displayed abnormal hemoglobin A1C levels at 5.9% (reference range <=5.6%) and estimated average glucose levels of 191 mg/dL (70 - 180 mg/dL). Following initiation of semaglutide treatment, these levels normalized during her latest assessment. EGBP at age 41, prompted by her history of hypokalemia and polyuria, initially yielded negative results. However, re-analysis of exome raw data revealed a likely pathogenic variant in HNF1A (NM_000545.8) c.1745A>G, p.(His582Arg) not previously assessed in the nephrology-focused EGBP. This variant is present in 13 alleles out of 240,596 in gnomAD and it is predicted deleterious (REVEL=0.69). It has been previously identified with suboptimal function in in vitro assays, and classified as a strong type 2 diabetes risk modifier in maturity-onset diabetes of the young (MODY) studies [18, 19]. This information was conveyed to the clinical team for further exploration of her diabetes diagnosis and assessment of the variant's significance in her family's diabetes history through segregation studies.
Comments (0)